ORIGINAL ARTICLE
Does the Inhibitory Action of Asymmetric Dimethylarginine (ADMA) on the Endothelial Nitric Oxide Synthase Activity Explain Its Importance in the Cardiovascular System? The ADMA Paradox
Dimitrios Tsikas
Core Unit Proteomics, Hannover Medical School, Hannover, Germany
Abstract
Asymmetric dimethylarginine (ADMA, NG, NG-dimethyl-L-arginine) is endogenously produced by asymmetric dimethylation of the guanidine group of L-arginine residues. ADMA is generally considered a powerful cardiovascular risk factor, an Übermarker, due to its inhibitory action on the activity of nitric oxide synthase (NOS) isoforms. In the endothelium, the constitutively expressed and Ca2+/calmodulin-dependent NOS (eNOS) catalyzes the conversion of L-arginine to nitric oxide (NO). NO is one of the most potent endogenous activators of soluble guanylyl cyclase which produces the second messenger cyclic guanosine monophosphate (cGMP). There is experimental evidence from in vitro and in vivo experiments that challenges the extraordinary importance of ADMA as the culprit of NO-related cardiovascular diseases in the human circulation. Most notably, we present data showing that ADMA is a weak competitive inhibitor of recombinant endothelial NOS (eNOS) activity (Ki 3.9 μM, IC50 12 μM). Thus, at its relatively low concentrations of 0.4 to 0.5 μM in the human blood, ADMA is unlikely to inhibit NO synthesis in the endothelium to an extent sufficient enough to cause endothelium dysfunction. Furthermore, ADMA does not “uncouple” eNOS and does not diminish the bioavailability of NO through its reaction with superoxide radical anions produced by “uncoupled” eNOS. Consequently, the particular importance assigned to ADMA in the human circulation is likely to be due to other not yet recognized biological actions beyond inhibition of eNOS activity. This “ADMA paradox” remains to be solved.
Keywords: L-arginine paradox; asymmetric dimethylarginine; cardiovascular disease; endothelial nitric oxide synthase; nitric oxide
Received: 17 May 2017;
Accepted after revision: 27 June 2017;
Published: 21 July 2017.
Author for correspondence: Dimitrios Tsikas, Core Unit proteomics, Hannover Medical School, Carl-Neuberg-Strasse 1, DE-30625 Hannover, Germany. Email:
[email protected]How to cite: Tsikas D. Does the Inhibitory Action of Asymmetric Dimethylarginine (ADMA) on the Endothelial Nitric Oxide Synthase Activity Explain Its Importance in the Cardiovascular System? The ADMA Paradox. J Controversies Biomed Res 2017;3(1):16–22.
DOI:
http://dx.doi.org/10.15586/jcbmr.2017.22Copyright: Tsikas D
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0
Introduction
Nitric oxide (NO) is considered to play a critical role in signal transduction pathways in the cardiovascular and nervous systems and is a key component of the cytostatic or cytotoxic function of the immune system (1, 2). In various pathological conditions associated with endothelial dysfunction, such as atherosclerosis, hypercholesterolemia, and essential hypertension, urinary excretion of the NO metabolites nitrate and nitrite, as well as of cGMP, the second messenger of NO, are reduced both in animals and in humans [discussed in Ref. (3)]. In these conditions, the plasma concentration of L-arginine, the substrate of nitric oxide synthase (NOS; EC 1.14.13.39) isoforms (4), is almost normal, while the plasma concentration of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NOS, is slightly increased. Intravenous or oral administration of L-arginine to healthy and diseased humans as well as to animals results in enhanced endothelium-dependent vasodilation, increase of plasma nitrite concentration and enhanced urinary excretion of nitrate and cGMP (3), which is indicative of enhanced NO synthesis. From a biochemical point of view, this is a surprising finding, because L-arginine is found in human blood at concentrations being approximately 10-fold higher than the KM values for all NOS isoforms (3). This “abnormality” is generally known as “The L-Arginine Paradox.”
Basal inhibition of NOS activity by ADMA is one possible explanation of this phenomenon. Known NOS isoforms include endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS) (5). All NOS isoforms including eNOS are known to produce not only NO but also the superoxide radical anion. NO produced by NOS and superoxide produced by NOS and/or other enzymes can lead to irreversible formation of peroxynitrite (ONOO–), a potent oxidant of various biomolecules including NOS. This reaction is considered particularly important because it diminishes NO bioavailability. Besides ADMA, there are additional endogenous and exogenous NG-substituted L-arginine inhibitors of NOS activity (1, 6–19). Yet, only ADMA has been suggested as a potent risk factor for the development of NO-associated endothelial dysfunction and cardiovascular diseases in humans due to its inhibitory action on eNOS (20, 21). However, there is a body of experimental and clinical evidence that argue against this widely widespread belief, notably in the cardiovascular system. The present study provides new experimental evidence for the weak inhibitory action of ADMA on eNOS and discusses the controversial issue and suggests the use of the expression “The ADMA Paradox” to characterize this phenomenon.
Materials and methods
Chemicals, enzymes, and reagents
Na15NO2 (98% at 15N) and L-[guanidino-15N2]-arginine hydrochloride (98% at both 15N atoms) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA). NaNO2 and NaNO3 were bought from Riedel de Haen (Seelze, Germany). 2,3,4,5,6-Pentafluorobenzyl (PFB) bromide was obtained from Aldrich (Steinheim, Germany). Toluene and acetone were purchased from Baker (Deventer, the Netherlands). Na15NO3 (98% at 15N), ADMA, L-NG-nitro-arginine (L-NNA), L-NG-methyl-arginine (L-NMA), NADPH, H4B, FAD, and FMN were obtained from Sigma (Munich, Germany). Calmodulin was bought from Biomol (Hamburg, Germany). CaCl2 and KH2PO4 were purchased from Merck (Darmstadt, Germany). Dilutions of NADPH, H4B, FAD, FMN, L-[guanidino-15N2]-arginine and all NOS inhibitors were freshly prepared in distilled water. Calmodulin (1 mg) was diluted in distilled water (1 mL), aliquoted, and stored frozen. Purified recombinant bovine eNOS (Cayman Chemical, Ann Arbor, MI) was isolated from a Baculovirus overexpression system in Sf9 cells and was supplied in 50 mM HEPES, pH 7.4, with 10% glycerol, 5 mM CHAPS, and 100 μM dithiothreitol (DTT). The specific activity of eNOS was declared by the supplier as 1.3 nmol NO/min × mg as measured spectrophotometrically using the oxyhemoglobin assay (1 mM CaCl2, 1.2 μM calmodulin, 0.1 mM NADPH, 50 μM L-arginine, 12 μM H4B, 170 μM DTT). The original eNOS preparation (1.07 mL, 6.18 mg eNOS) was thawed, aliquoted to 10–μL portions, and frozen (–80°C) immediately. eNOS stock solutions were used once without re-refrigerating.
Measurement of recombinant bovine eNOS activity
eNOS assays were performed with minor modifications of the GC–MS method recently developed for and applied to nNOS (19). Fresh solutions of NOS cofactors were prepared daily in potassium phosphate buffer (50 mM, pH 7, 5 mL), resulting in the following final concentrations: H4B, 10 μM; FAD, 5 μM; FMN, 5 μM; CaCl2, 500 μM; NADPH, 0.8 mM; calmodulin, 500 nM. In experiments on eNOS inhibition, NOS inhibitors were added at final concentrations of 1, 2, 5, or 10 μM (see below). Aliquots (400 μL) of these solutions were spiked with varying concentrations of L-[guanidino-15N2]-arginine (range 0–40 μM) and pre-incubated for 10 min at 37°C. The reaction was started by addition of eNOS at a final protein concentration ranging between 5 and 90 μg/mL. The reaction was stopped after various times of incubation (range 0–30 min) by transferring aliquots (100 μL) of eNOS incubates into 1.5-mL glass vials, which contained an ice-cold acetonic solution (400 μL) of 14NO2− and 14NO3− for use as internal standards, resulting in a final concentration of 1.0 μM each. Samples were mixed immediately by vortexing and put on ice until derivatization by the following procedure. PFB bromide (10 μL) was added and samples were derivatized by heating at 50°C for 60 min. After cooling to room temperature, acetone was removed under nitrogen and reaction products were extracted with toluene (300 μL) by vortex-mixing for 1 min. GC–MS analyses of 0.5-μL aliquots were performed in the negative-ion chemical ionization mode on a Hewlett-Packard MS Engine 5989A connected directly to a gas chromatograph 5890 series II (Waldbronn, Germany), which was equipped with a fused-silica capillary column Optima 17 (15 m × 0.25 mm I.D., 0.25-μm film thickness) from Macherey-Nagel (Düren, Germany). Selected ion monitoring of the ions with a mass-to-charge ratio (m/z) of m/z 47 (15NO2–) for [15N]nitrite, m/z 46 (14NO2–) for [14N]nitrite, m/z 63 (15NO3–) for [15N]nitrate, and m/z 62 (14NO3–) for [14N]nitrate was performed as described (19).
Investigations of the inhibitory mechanism of ADMA on eNOS
Cofactor concentrations were as described above. L-[guanidino-15N2]-arginine was used at fixed concentrations of 0, 2.5, 5, 7.5, 10, 12.5, 15, 20, 30, and 40 μM. ADMA was used at 0 and 5 μM (each in triplicate), and at 1, 2, 10 μM (each in quadruplicate). eNOS concentration, incubation temperature, and incubation time were 20.2 μg/mL, 37°C, and 10 min, respectively. Samples were treated and analyzed by GC–MS as described above. Kinetics constants and type of inhibition were determined from Lineweaver–Burk and Hanes plots using mean-specific eNOS activities, which were determined from measured 15NO3– and 15NO2– concentrations and expressed as nmol 15NO3– or 15NO2– per min and per mg eNOS.
Results
The mechanisms by which ADMA inhibits NOS isoforms are incompletely understood. Moreover, many results arise from cell or in vivo experiments which by nature cannot provide information about the kind of inhibition. The IC50 value for ADMA in rat brain homogenates has been reported to be 2 μM (22). Our group found that ADMA is a potent non-competitive inhibitor (Ki 0.4 μM; Kii 1.6 μM; IC50 1.5 μM) of recombinant rat nNOS (3, 19, 23, 24). By contrast, others have reported that ADMA is a weak competitive inhibitor (Ki 0.7 μM; IC50 ≈ 20 μM) of rat nNOS (25). By means of a reliable GC-MS assay (19), we performed detailed mechanistic studies on the inhibitory action of ADMA on a commercially available recombinant bovine eNOS. The results of these studies are shown in Figures 1–3 and summarized in Tables 1–3.
Table 1. Statistical data from primary Lineweaver–Burk plots
ADMA
(μM) |
[15N]nitrite |
[15N]nitrate |
A
(SE) |
B
(SE) |
r
(SD) |
A
(SE) |
B
(SE) |
r
(SD) |
| 0 |
1.089 |
7.91 |
0.987 |
1.048 |
5.49 |
0.967 |
| |
(0.079) |
(0.48) |
(0.161) |
(0.09) |
(0.55) |
(0.183) |
| 1 |
1.014 |
11.0 |
0.993 |
0.906 |
6.28 |
0.962 |
| |
(0.084) |
(0.51) |
(0.170) |
(0.111) |
(0.68) |
(0.225) |
| 2 |
1.126 |
13.2 |
0.997 |
1.161 |
7.37 |
0.986 |
| |
(0.064) |
(0.4) |
(0.131) |
(0.078) |
(0.48) |
(0.159) |
| 5 |
1.079 |
18.1 |
0.998 |
1.066 |
11.5 |
0.991 |
| |
(0.081) |
(0.5) |
(0.156) |
(0.109) |
(0.6) |
(0.210) |
| 10 |
1.257 |
30.1 |
0.995 |
1.274 |
16.3 |
0.990 |
| |
(0.186) |
(1.1) |
(0.378) |
(0.146) |
(0.9) |
(0.296) |
| Y-Axis intercepts (A), slopes (B), and coefficients of correlation (r) values from primary Lineweaver-Burk plots for [15N]nitrite and [15N]nitrate production rates measured in the absence and in the presence of ADMA at the indicated concentrations. Values in parentheses represent the standard error (SE) for A and B or the standard deviation (SD) for r. |
Table 2. Statistical data from secondary Lineweaver–Burk and Hanes plots
A
(SE) |
[15N]nitrite |
[15N]nitrate |
B
(SE) |
r
(SD) |
A
(SE) |
B
(SE) |
r
(SD) |
| Secondary Lineweaver-Burk plot |
| 8.352 |
2.143 |
0.997 |
5.383 |
1.112 |
0.997 |
| (0.49) |
(0.097) |
(0.783) |
(0.25) |
(0.049) |
(0.395) |
| Secondary Hanes plot |
| 7.878 |
2.395 |
0.983 |
5.779 |
1.352 |
0.999 |
| (1.33) |
(0.260) |
(2.098) |
(0.14) |
(0.028) |
(0.222) |
| Y-Axis intercepts (A), slopes (B), and coefficients of correlation (r) values from secondary Lineweaver-Burk plots and Hanes plots for [15N]nitrite and [15N]nitrate production rates measured in the absence and in the presence of ADMA at the indicated concentrations. Values in parentheses represent the standard error (SE) for A and B or the standard deviation (SD) for r. |
Table 3. Michaelis–Menten constants for recombinant bovine eNOS
| ADMA (μM) |
[15N]nitrite |
[15N]nitrate |
Vmax
(nmol/min × mg) |
KM
(μM) |
Vmax
(nmol/min × mg) |
KM
(μM) |
| 0 |
0.918 |
7.26 |
0.954 |
5.24 |
| 1 |
0.986 |
13.6 |
1.104 |
8.37 |
| 2 |
0.888 |
17.8 |
0.861 |
8.97 |
| 5 |
0.927 |
38.7 |
0.938 |
22.0 |
| 10 |
0.796 |
85.3 |
0.785 |
39.3 |

Figure 1. Time dependency of the inhibition of eNOS activity by various guanidino–substituted L–arginine analogs. eNOS-dependent (45 μg/ml) formation of 15NO2– (A) and 15NO3– (B) from L–[guanidino–15N2]arginine (20 μM) in the absence of any inhibitor and in the presence of ADMA, L-NNA, or L-NMA (each at 1 μM). Values are shown as mean ± SD from duplicate determinations.

Figure 2. Primary (A and B) and secondary (C) Lineweaver-Burk plots for 15NO2– and 15NO3–. The experimental conditions are described in the “Materials and Methods” section.

Figure 3. Effects of intravenously administered ADMA at infusion rates of 0.0, 12.5, 25, 37.5, and 75 μg/kg/min in six healthy volunteers on plasma cGMP levels and ERPF. The corresponding ADMA peak plasma concentrations were 0.94 ± 0.26, 4.17 ± 0.99, 8.13 ± 2.08, 11.8 ± 2.53, and 15.7 ± 4.29 μM. Data are shown as mean ± SD. Asterisks indicate statistical significance (P < 0.05) between post-infusion and pre-infusion values. This figure was constructed using data reported previously by our group (26).
The mean specific activity of this eNOS (0.41 nmol 15NO3–/min × mg eNOS, and 0.48 nmol 15NO2–/min × mg eNOS) is about 300 times smaller than that of a recombinant rat nNOS (19), indicating that eNOS is much less active compared to nNOS. ADMA and L-NMA were found to be equipotent inhibitors of eNOS activity (Figure 1). A Michaelis–Menten kinetics was observed (Figure 2; Tables 1–3). ADMA was found to be a competitive inhibitor of eNOS activity. Vmax was determined to be 0.90 ± 0.07 nmol 15NO2–/min × mg eNOS and 0.92 ± 0.12 nmol 15NO3–/min × mg eNOS. The inhibition constant Ki for ADMA was determined to be 3.9 μM with respect to 15NO2– and 4.8 μM with respect to 15NO3–. At the highest concentration of ADMA used in these experiments, that is, 10 μM, ADMA inhibited eNOS activity by less than 50%, suggesting that the IC50 of ADMA for eNOS is above 10 μM, which was estimated to be approximately 12 μM.
We have previously reported that infusion of ADMA (up to 75 μg/kg/min for 40 min) in six healthy volunteers resulted in dose-dependent increases of plasma ADMA concentration, and in decreases of circulating cGMP levels (by 18%–27%) and effective renal plasma flow (EPRF; by 0.1%–7%) (26, 27) (Figure 3). These in vivo observations are in line with those obtained in vitro using eNOS in the present study.
Discussion
Formulas (1), (2), and (3) describe the theoretical extent of inhibition (EI, in %) by ADMA of eNOS and nNOS activity (V) for competitive and non-competitive mechanisms for ADMA concentrations (I) in the range 0–20 μM and for three constant L-arginine concentrations (S) of 50, 200, and 500 μM. In these expressions, V is the NOS activity in the presence of ADMA and V0 is the NOS activity in the absence of the ADMA. By means of these formulas and the experimental results, the EI values were calculated and plotted versus the ADMA concentration (Figure 4). In this figure, the extent of inhibition of plasma cGMP and of ERPF upon infusion of ADMA in six healthy volunteers versus the corresponding peak plasma concentration of ADMA (see Figure 3) is also plotted.
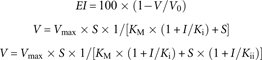

Figure 4. Calculated (curves) dependence of the extent of inhibition of eNOS and nNOS by ADMA (0– 20 μM) at three fixed concentrations of L-arginine (i.e., 50, 200, and 500 μM). The extent of inhibition was calculated by using the experimentally observed Michaelis–Menten kinetics constants in the present study for eNOS and in previous work for nNOS (KM = 6 μM for eNOS and 3 μM for nNOS; Ki = 4 μM for eNOS and 0.4 μM for nNOS; Kii = 1.6 μM for nNOS) (3, 23, 24). Open squares and circles are experimentally observed data on the extent of inhibition of plasma cGMP and ERPF upon infusion of ADMA and are indicated by arrows.
Figure 4 shows that nNOS activity is potently inhibited by ADMA, even at high pharmacologically relevant concentrations of the NOS substrate L-arginine. ADMA at 10 μM inhibits nNOS activity by 90%. At pathophysiological plasma concentrations of ADMA, that is, 0.5 to 1 μM (28), nNOS activity would be inhibited by approximately 30%. Because of the non-competitive inhibitory mechanism, nNOS activity can be increased only by decreasing intracellular ADMA concentration, for instance, by supplementing high amounts of L-arginine.
In contrast, eNOS activity is weakly and competitively inhibited by ADMA (Figure 4). Thus, even at very high ADMA concentration of 10 μM, eNOS activity would be inhibited by only 22%. At physiological plasma concentrations of ADMA, eNOS activity would be inhibited by less than 5%. Doubling the ADMA plasma concentration to reach pathologically elevated ADMA levels will not markedly increase the extent of inhibition of eNOS activity. An increase in L-arginine concentration from the physiological plasma concentration of about 50 μM to a pharmacological level of 500 μM would have a much stronger effect on eNOS activity and would almost completely abolish the inhibition of eNOS activity by ADMA.
On the assumption that the in vitro observations of our group on the inhibition of isolated eNOS and nNOS by ADMA are valid and extendable to humans and animals in vivo, one can assume that under physiological conditions all NOS isoforms will be partially inhibited by ADMA, with nNOS activity being much stronger inhibited than eNOS activity. With respect to the NOS activity in endothelial cells, eNOS activity would increase both through competition of administered L-arginine with ADMA and by exchanging intracellular ADMA by extracellular L-arginine (Figure 4). Based on the different mechanisms of action of ADMA on eNOS and nNOS, it may be expected that supplementation of L-arginine will reduce extent of basal inhibition of NOS activity by ADMA by the same degree in endothelial and neuronal cells (Figure 4). These considerations led to conclude that administration of relatively large amounts of L-arginine to humans will produce the same beneficial NO-related effects in the whole human organism.
Conclusion
In vitro, ADMA is a weak competitive inhibitor of eNOS activity in vitro, but is a relatively strong non-competitive inhibitor of nNOS. ADMA infused to healthy subjects exerts only very weak effects in the renal and cardiovascular systems, even at plasma concentrations that are about 10 times higher than those measured in many pathological conditions. It can be concluded that ADMA is unlikely to be a predominant determinant of the eNOS activity in the human circulation. The cardiovascular risk assigned to ADMA in adults is likely to arise from other not yet recognized actions of ADMA. Children have much higher circulating ADMA concentrations than adults (29), but there is no evidence that children have a risk for cardiovascular disease. In humans, the activity of eNOS, nNOS, and iNOS is steadily inhibited by ADMA and other endogenous inhibitors, and L-arginine administration can reduce the extent of this baseline inhibition to almost the same degree, albeit by different mechanisms. This is a likely explanation of the “L-Arginine Paradox.” The very weak inhibitory potency of ADMA on eNOS activity in the human endothelium does not support the cardiovascular risk ascribed to ADMA thus far. We name this phenomenon “The ADMA Paradox” which is presently unexplored and remains to be resolved.
Conflicts of interest
The authors report no conflicts of interest with respect to research, authorship, and/or publication of this article.
References
- Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–64.
- Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies. FASEB J. 1995;9:1319–30.
- Tsikas D, Böger RH, Sandmann J, Bode-Böger SM, Frölich JC. Endogenous nitric oxide synthase inhibitors are responsible for the L-arginine paradox. FEBS Lett. 2000;478:1–3. http://dx.doi.org/10.1016/S0014-5793(00)01686-0
- Marletta MA. Nitric oxide synthase structure and mechanism. J Biol Chem. 1993;268:12231–4.
- Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–31. http://dx.doi.org/10.1016/S0008-6363(99)00115-7
- Hibbs JB, Taintor RR, Vavrin Z. Macrophage cytotoxicity: Role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science. 1987;235:473–6. http://dx.doi.org/10.1126/science.2432665
- Rees DD, Palmer RMJ, Hodson HF, Moncada S. A specific inhibitor of nitric oxide formation from L-arginine attenuates endothelium-dependent relaxation. Br J Pharmacol. 1989;96:418–24. http://dx.doi.org/10.1111/j.1476-5381.1989.tb11833.x
- Vallance P, Leone A, Calver A, Collierr J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992;339:572–5. http://dx.doi.org/10.1016/0140-6736(92)90865-Z
- Kotani K, Ueno SI, Kakimoto Y. Isolation and identification of methylarginines from bovine brain. J Neurochem. 1992;58:1127–9. http://dx.doi.org/10.1111/j.1471-4159.1992.tb09371.x
- Bredt DS, Snyder SS. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–5. http://dx.doi.org/10.1073/pnas.87.2.682
- Klatt P, Schmidt K, Uray G, Mayer B. Multiple catalytic functions of brain nitric oxide synthase. J Biol Chem. 1993;268:14781–7.
- Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–17. http://dx.doi.org/10.1021/bi00084a017
- Klatt P, Schmidt K, Brunner F, Mayer B. Inhibitors of brain nitric oxide synthase: Binding kinetics, metabolism, and enzyme inactivation. J Biol Chem. 1994;269:1674–80.
- Furfine ES, Carbine K, Bunker S, Tanoury G, Harmon M, Laubach V, Sherman P. Potent inhibition of human neuronal nitric oxide synthase by NG-nitro-L-arginine methyl ester results from contaminating NG-nitro-L-arginine. Life Sci. 1997;60:1803–9. http://dx.doi.org/10.1016/S0024-3205(97)00140-9
- Kielstein A, Tsikas D, Galloway GP, Mendelson JE. The inhibitory action of ADMA and its importance in the cardiovascular. Nitric Oxide. 2007;17:55–9.
- Olken NM, Rusche KM, Richards MK, Marletta MA. Inactivation of macrophage nitric oxide synthase by NG-methyl-L-arginine. Biochem Biophys Res Commun. 1991;177:828–33. http://dx.doi.org/10.1016/0006-291X(91)91864-9
- Pufahl RA, Nanjappan PG, Woodard RW, Marletta MA. Mechanistic probes of N-hydroxylation of L-arginine by the inducible nitric oxide synthase from murine macrophages. Biochemistry. 1992;31:6822–8. http://dx.doi.org/10.1021/bi00144a024
- Feldmann PL, Griffith OW, Hong H, Stuehr DJ. Irreversible inactivation of macrophage and brain nitric oxide synthase by L-NG-methylarginine requires NADPH-dependent hydroxylation. J Med Chem. 1993;36:491–6. http://dx.doi.org/10.1021/jm00056a009
- Tsikas D, Sandmann J, Savva A, Lueßen P, Böger RH, Gutzki FM, et al. Assessment of nitric oxide synthase activity in vitro and in vivo by gas chromatography-mass spectrometry. J Chromatogr B. 2000;742:143–53. http://dx.doi.org/10.1016/S0378-4347(00)00142-0
- Böger RH, Bode-Böger SM, Szuda A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine: A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation. 1998;98:1842–7. http://dx.doi.org/10.1161/01.CIR.98.18.1842
- Leiper J, Vallance P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res. 1999;43:542–8. http://dx.doi.org/10.1016/S0008-6363(99)00162-5
- Faraci FM, Brain JE Jr, Heistad DD. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol. 1995;269:H1522–7.
- Sandmann J. PhD thesis, Untersuchungen zur Bedeutung der S-Transnitrosylierungs-Reaktion für die Bildung, den Metabolismus und die Vermittlung der biologischen Aktivitäten von NO und S-Nitroso-Substanzen. University of Hannover, Hannover, Germany, 2002.
- Albsmeier J. PhD thesis, Bedeutung endogener Inhibitoren der NO-Synthase für kardiovaskuläre Erkrankungen und ihre pharmakologische Therapie. University of Hamburg, Hamburg, Germany, 2004.
- Cardounel AJ, Zweier JL. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J Biol Chem. 2002;277:33995–4002. http://dx.doi.org/10.1074/jbc.M108983200
- Kielstein JT, Impraim B, Simmel S, Bode-Böger SM, Tsikas D, Frölich JC, et al. Cardiovascular effects of systemic NO synthase inhibition with asymmetric dimethylarginine in humans. Circulation. 2004;109(2):172–7. http://dx.doi.org/10.1161/01.CIR.0000105764.22626.B1
- Kielstein JT, Tsikas D, Fliser D. Effects of asymmetric dimethylarginine (ADMA) infusion in humans. Eur J Clin Pharmacol. 2006;62(Suppl 2):39–44. http://dx.doi.org/10.1007/s00228-005-0010-1
- Tsikas D, Junker W, Frölich JC. Determination of dimethylated arginines in human plasma by high-performance liquid chromatography. J Chromatogr B. 1998;705:174–6.
- Lücke T, Kanzelmeyer N, Kemper MJ, Tsikas D, Das AM. Developmental changes in the L-arginine/nitric oxide pathway from infancy to adulthood: Plasma asymmetric dimethylarginine levels decrease with age. Clin Chem Lab Med. 2007;45(11):1525–30. http://dx.doi.org/10.1515/CCLM.2007.300



