|
|
||||||||||||
|
Free Neuropathology 4:12 (2023) |
||||||||||||
|
Flashback |
||||||||||||
|
From amaurotic idiocy to biochemically defined lipid storage diseases: the first identification of GM1-Gangliosidosis |
||||||||||||
|
Burkhard S. Kasper1, Christian Thomas2, Anne Albers2, Ekkehard M. Kasper3, Konrad Sandhoff4 |
||||||||||||
|
||||||||||||
|
Corresponding author: |
||||||||||||
|
Additional resources and electronic supplementary material: supplementary material |
||||||||||||
|
Submitted: 22 May 2023 |
||||||||||||
|
Keywords: Lipid storage disease, GM1-gangliosidosis, Amaurotic idiocy, Tay-Sachs-Disease, Beta-galactosidase |
||||||||||||
|
Original paper: On a biochemically special form of infantile amaurotic idiocy. Jatzkewitz H., Sandhoff K., Biochim. Biophys. Acta 1963; 70; 354-356. See supplement 1. |
||||||||||||
|
Abstract On February 23rd 1936, a boy-child (“Kn”) died in an asylum near Munich after years of severe congenital disease, which had profoundly impaired his development leading to inability to walk, talk and see as well as to severe epilepsy. While a diagnosis of “Little’s disease” was made during life, his postmortem brain investigation at Munich neuropathology (“Deutsche Forschungsanstalt für Psychiatrie”) revealed the diagnosis of “amaurotic idiocy” (AI). AI, as exemplified by Tay-Sachs-Disease (TSD), back then was not yet understood as a specific inborn error of metabolism encompassing several disease entities. Many neuropathological studies were performed on AI, but the underlying processes could only be revealed by new scientific techniques such as biochemical analysis of nervous tissue, deciphering AI as nervous system lipid storage diseases, e.g. GM2-gangliosidosis. In 1963, Sandhoff & Jatzkewitz published an article on a “biochemically special form of AI” reporting striking differences when comparing their biochemical observations of hallmark features of TSD to tissue composition in a single case: the boy Kn. This was the first description of “GM1-Gangliosidosis”, later understood as resulting from genetically determined deficiency in beta-galactosidase. Here we present illustrative materials from this historic patient, including selected diagnostic slides from the case “Kn” in virtual microscopy, original records and other illustrative material available. Finally, we present results from genetic analysis performed on archived tissue proving beta-galactosidase-gene mutation, verifying the 1963 interpretation as correct. This synopsis shall give a first-hand impression of this milestone finding in neuropathology. |
||||||||||||
|
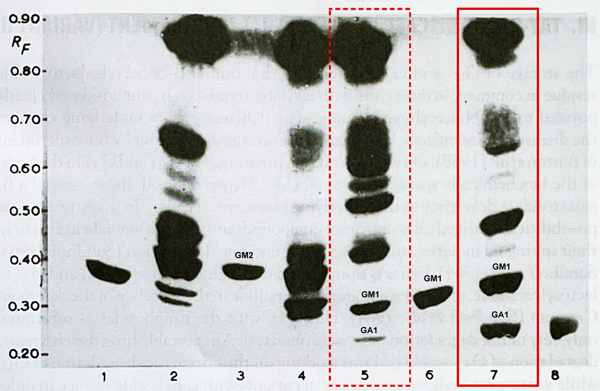
Introduction In history of science, there has always been and will be discussion, to whom to ascribe to the first description of a disease. Addressing a specific publication as the first description can turn out quite differently, depending on whether or not one is focusing on a disease’s clinical manifestations, its tissue pathology, pathophysiological mechanism or underlying genetic cause. Further complicating matters, similar findings are sometimes reported independently or even simultaneously by separate researchers at different places. As thinking and knowledge about human disease evolve over time, which include terminology and classifications, it could well be that we end up with more than one author credited for the first description of a particular disease. Individual patients and the details behind groundbreaking findings, however, are rarely appreciated in detail, albeit some of them have become linked to (neuro-)history at least by their initials, such as “H.M.” [1]. Concerning the “lipidoses”, exemplified by Tay-Sachs disease (TSD), syndromal appearance of certain symptoms in children had been well observed and reported as early as at the end of the 19th century by Warren Tay [55] and Bernard Sachs [36] establishing “Tay-Sachs-Disease” as an eponym [6], as well as “amaurotic family idiocy” (AFI) as another common term [35]. The rather dramatic clinical presentation in TSD/AFI and comparable diseases, such as Niemann-Pick’s, was accompanied by striking pathologic findings post-mortem, especially in the CNS, where neurons were found enlarged and ballooned, obviously filled by aberrant material [25]. The first pathoanatomic description of TSD appears to have been given by Sachs himself in 1887 [36]. In the 20th century TSD/AFI brains were studied extensively both histopathologically and later also ultrastructurally (e.g. [8], [9], [10], [38], [55], [56]) laying the foundation for a concept of lysosomal lipid storage disease (review see [11], [25], [27], [58]). CNS tissue pathology in TSD and other storage diseases appeared rather uniform, resulting from accumulation of some storage material strikingly altering neuronal morphology, a finding early appreciated in CNS histopathology ([12], [57], [46-48]). AFI subtypes for quite a long time were separated on clinical grounds only by predominating manifestation age. Specific techniques allowing a more precise analysis, deciphering the underlying molecular pathobiology and establishing a clear understanding of different disease forms, were not available. This changed in the mid-to-late 1930s, when Klenk coined the term “gangliosides” to describe characteristic acid glycolipid components of neuronal cells isolated from TSD brains [19-21]. It took more than 20 years from then to identify the first ganglioside structure G1 [24], now called GM1, and to apply better techniques such as thin-layer chromatography allowing for separation and characterization of the different ganglioside components in diseased versus normal brain tissues in the 1960s. Further important breakthrough discoveries were achieved in the understanding of nervous system within a few years ([14], [15], [24], [34], [43], [53]) deciphering the “gangliosidoses”, including classic TSD as GM2-gangliosidosis. Research activities and knowledge gain then exploded, leading to one of the most astonishing advances in understanding of lipid storage-related CNS-disease (for review see: [2], [16]), which for decades before had remained enigmatic and summed up under different terms of AFI, “amaurotic idiocy” (AI) or “infantile amaurotic idiocy” (IAI), a hot topic in early neuropathology. Up to today, many aspects of lipid storage related disease mechanisms have been elucidated. This includes many of the enzymes involved, their functional dynamics in health and disease, their structure(s), co-factors and substrates including cellular pathways & regulation, the role of the lysosomal compartment, the regulating genes and their mutational spectrum (for review see [2], [16], [22], [28], [42]). After decades characterized by restriction to symptom-oriented diagnosis and, at best, post mortem validation in a few selected cases, we are now able to recognize these diseases intra vitam by surrogate biomarkers and/or molecular testing. It is a fascinating journey to follow the historic lines of these developments. Here we present and re-appreciate a key patient, “Kn”, including his post mortem tissues as well as the understanding derived from analyzing his tissue probes biochemically: this boy died 1936 in Munich from a fatal course of IAI at the age of 7¾. By diligent biochemical analysis of this patient’s brain tissue, performed after more than 25 years of formalin-fixation, Jatzkewitz & Sandhoff revealed a “biochemically special form of infantile amaurotic idiocy” [14] later to be named “GM1-gangliosidosis” [51]. This can be acknowledged as the first definite characterization of this disease [16, 28], eliciting that TSD/AI/IAI was a spectrum of different diseases rather than a uniform entity. What exactly did the authors describe in this study? Jatzkewitz & Sandhoff presented a surprising result of a thin-layer-chromatographic lipid compound analysis performed on brain tissue from an “AI”-patient, for this single case (patient “Kn”) out of an AI-series clearly displayed an unusual “aberrant” pattern on chromatography when compared to chromatography bands of other “AI-TSD”-materials and normal brain (see Figure 1). Kn’s analysis did not show the accumulation of “Tay-Sachs-ganglioside” (=GM2 ganglioside) but showed accumulation of the major normal “ganglioside G1” according to Kuhn & Wiegandt [24], i.e. ganglioside A according to Klenk and GM1-ganglioside according to Svennerholm. Why is this a milestone paper? The paper’s key finding - summarized on less than three pages - convincingly demonstrates that a special and biologically different variant of TSD-like disease had been found, which turned out to be correct. In contrast to classic TSD showing accumulation of GM2-ganglioside and its sialic acid free residue GA2, here a different metabolic block had to be suggested since other substances were stored, identified as “ganglioside GM1” and its sialic acid free residue [14] [41], which turned out to be correct [2]. The distinct identity of these storage compounds was proven by chemical, biochemical and enzymatic procedures [43]. While several papers and textbooks refer to a different study as the one having identified GM1 as the storage compound in this type of disease [30], this clearly came later ([16], [28]). At this stage, all the knowledge to be revealed later about metabolic chains and enzymes involved had not yet been known [44].
Figure 1. Original chromatography analysis by Sandhoff: “Kn” = lane 7. Formalin-fixed control = lane 5 (Photography by K. Sandhoff, gangliosides labelled; original legend). See full article by Jatzkewitz & Sandhoff 1963 [14] in supplement 1 (illustrating the metabolic path).Thin-layer chromatogram of lipid extracts of brain tissue from two usual infantile cases and one from one special form of late-infantile amaurotic idiocy (Jatzkewitz and Sandhoff 1963) Adsorbent, 400-µm-thick layer of Kieselgel G, Merck; solvent system, propanol-conc. ammonia-water (6:2:1); height of the solvent front, 15cm; detection, anisaldehyde, sulfuric acid in acetic acid (reagent of Kägi-Miescher). 1, 20µg of neuraminic acid-free residue of ganglioside Tay-Sachs (B’, now named GA2, RF 0.33); 2, 250µg of total lipid extract of the brain cortex of a case of iai (fresh tissue); 3, 20µg of ganglioside Tay-Sachs (A’, GM2, RF 0.37); 4, 250µg of total lipid extract of the brain cortex of a case of iai, preserved for formalin for 26 years; 5, 250µg of total lipid extract of normal brain cortex, preserved in formalin for 26 years; 6, 20µg of ganglioside A (GM1, RF 0.30); 7, 250µg of total lipid extract of the brain cortex of a special form of late iai, preserved in formalin for 26 years; 8, 20µg of neuraminic acid-free residue of ganglioside A (GA1, RF 0.26). [Author’s addendum: RF means “Retention factor”]. What exactly was new based on the knowledge back then? Up to this 1963 study’s result, AI and TSD was perceived as an entity with varying clinical courses and was classified according to manifestation age. Classic microscopic histomorphological analysis was not able to clearly differentiate between various entities, and therefore could not resolve the AI-enigma, maybe except separation of the “Neuronal Ceroid-lipofuscinoses” (NCL) as a separate group in AI-like disease ([46-48] [57]). Of note, NCL as a term was coined later ([50],[62]). The biology and biochemical nature of these diseases were largely unknown. What turned out as being correct or incorrect since then? Some authorities were convinced that essentially everything was already known about TSD up to 1960. As late as 1957, Seitelberger had stated that “…it has been…proven, that the cellular metabolic disturbance constituting the individual forms [of TSD] are mainly the same” [49]. When K. Sandhoff - following his analysis of a randomly selected first AI-case - searched for further brain specimens in Munich neuropathology archives, he was confronted by the institute’s head G. Peters, who told him boldly: “…if you aim to know about Tay-Sachs-disease, read my book and you will know everything” (Sandhoff, personal memory). This was the starting point. Newly developed biochemical analytic methods opened new windows: it turned out that diverse lipid compounds represent essential building blocks of nervous tissue, that their metabolism is highly sophisticated biochemically and can be altered causing various specific diseases (for review see [45], [2]). Concerning the “biochemically special form” of AI as identified in 1963, the postulated metabolic block in ganglioside A degradation later was identified and characterized as impairment of specific enzyme activities, mostly beta-galactosidase ([2], [16], [44]). At what stage of the author´s career was the paper published? K. Sandhoff was a young student at the very beginnings of his career as a researcher (Figure 2).
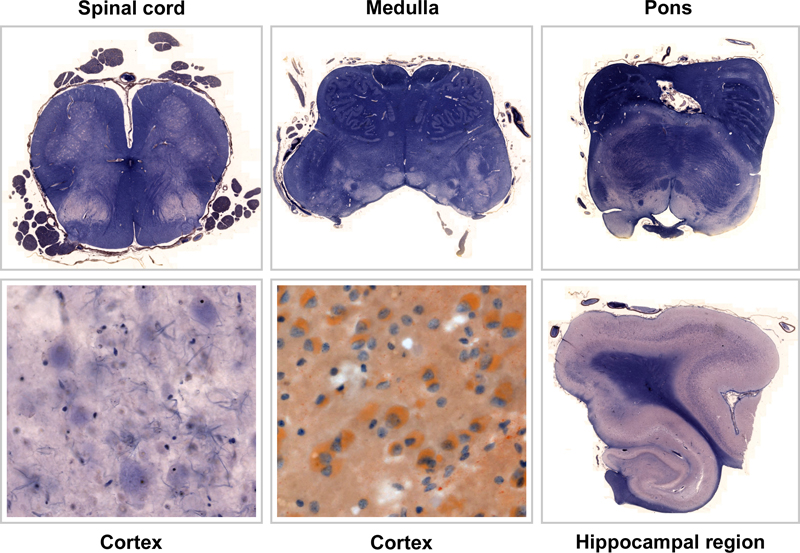
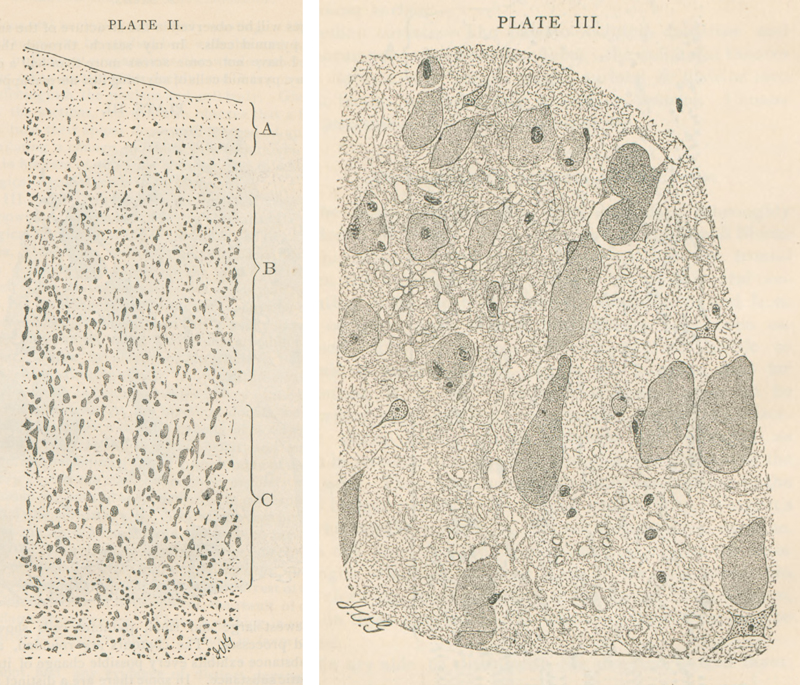
Figure 2: Konrad Sandhoff 1962, during the time of his thesis (Photo shared by K Sandhoff). This study was his debut publication. Having finished studying chemistry in 1962, he turned towards lipid compound analysis from nervous tissue by joining the department of neurochemistry headed by Horst Jatzkewitz at Munich Max-Planck Institute for Psychiatry, encouraged and supported by 1964 Nobel laureate Prof. Fedor Lynen. After failed attempts to isolate presumed sialic-acid-free gangliosides (“asialo-gangliosides”) from cattle brain, his attention turned towards new targets for investigation: rather by chance, in 1962 he decided to choose a first human brain sample in the Lab labelled “AI” [44]. In this very first analysis, using chromatographic techniques, he was able to reveal two bands, which he named “A” and “B” (later identified as GM2-ganglioside and its corresponding asialo-residue GA2). In order to validate his finding, he widened his analysis to include tissue probes from other 12 brains with AI/TSD picked from the archives at Munich neuropathology, “Kn” being one of them ([43], [44]). How was the paper received over time? Issues were raised whether the results presented were rather a methodological artifact caused by long-term formalin fixation, although this was largely ruled out by the experiments presented, since matching long-term fixed control samples had been included ([14], see Figure 1). In the realm of Anglo-American work this type of lipidosis became rather known as Norman-Landing-Disease or Landing’s disease for - after an early case reported by Norman [29] - Landing published a clinical report in 1964 ([26], [37], which was followed by identification of GM1-storage [30] and galactosidase deficiency [31]. In one early review, O’Brien questioned, whether the patient from the Jatzkewitz-Sandhoff-paper had the disease at all [30]. The Jatzkewitz-Sandhoff paper [14] prompted further group-activities supported by national and international colleagues, who agreed to provide selected tissues probes incl. fresh and fresh-frozen samples from “AI” patients for further analysis by Sandhoff. Several researchers knew each other personally and also cooperated, such as Sandhoff and O’Brien or Sandhoff and H. Moser/Boston (Sandhoff, personal communication, [39]). Such cooperation rapidly expanded the experience and led to description of further lipid storage disease variants, especially with respect to the GM2-gangliosidoses, including the hexosaminidase-0-variant later known as “Sandhoff’s disease” ([23], [33], [40]). Beyond this, further variants were described, including cases with storage but normal enzyme levels, indicating the presence of co-factors [44]. What happened to the original histological slides and patient documentation until today? Kn’s original slides were found within the Historical Archive of the Max Planck Institute for Psychiatry in Munich by registrar C. Dücker after intense search by BS Kasper had revealed enough detail in order to clearly identify them correctly, for there was no specific finding aid. The crucial informations were patient initials (“Kn”) and age (“7¾“), as well as the institutional archive case number (55/36) listed in the Sandhoff doctoral thesis only ([43], see supplement 2). An earlier paper on AI from Munich [8], which had recruited its cases from the Munich neuropathology collection also, served as cross reference, since it happened to list respective overlapping information including patient initials and age (see supplement 3). Thus, tissue samples of “case 55/36” could be identified in two case boxes labelled “AI” at MPI archives (see supplement 4). Luckily, the MPI archive stored one single additional document relating to “Kn”, a clinical report giving some more details about the patient child, including full name, date of birth, date of death and the name of the caring medical institution prior to his death (see supplement 5). This finally allowed to identify further clinical records preserved within another Munich archive, i.e. the Archbishop of Munich & Freising’s archive (see supplement 6); for reasons of clerical law and tradition, this archive keeps documents much longer than done by average public hospitals. The Munich Psychiatric hospital’s Archive did no longer store any documents (explored by P. Rauh, see acknowledgement). What is known about the patient behind the paper? The patient designated “Kn” in the respective publications ([8], [14], [15]) was a boy named Johann Knott, born April 11th 1928. Early on, he lived with a foster mother in Munich, but soon was put under guardianship by Munich city administration. This way he came into an asylum in Schönbrunn near Munich early due to his severe disability labelled “Little’s disease”. Asylum notes describe the kid as unable to talk, unable to walk, suffering from epilepsy and being “very poor”. He was referred to the Munich University Hospital (“Nervenklinik”) in June 1932, where he stayed until April 1933. At admission he was described as apathic and without reaction to external stimuli. He was noted to show impaired swallowing and an aberrant breathing pattern. Physical and neurological examination was reported with brisk reflexes, abnormal muscular tone, but ocular fundi were reported as normal. The boy had multiple epileptic seizures every day described as tonic without further detail. Affected by pneumonia, he died at Schönbrunn February 23rd 1936 (see supplements 5-7). It is important to note, that Kn’s death and postmortem investigation in no way is related to the Nazi euthanasia program (confirmation with support by P. Rauh; see acknowledgement, please read supplement 10). The role of German Neuropathology as well as clinical medicine during the NS-regime is at the focus of ongoing research (see supplement 10). What can be seen on the original slides? At the historical archive of the Munich Max-Planck-Institute for Psychiatry, 194 microscopic slides were found stored together with the original diagnostic report (see supplements 4 & 9) and the clinical note mentioned above (supplement 5). The slides encompass a systematic selection of CNS sites including specimens of the spinal cord, selected brain stem levels such as medulla and pons, cerebellum and various cortical samples quite differently stained. Stains according to the original report and (inconstant) slide labelling included Bielschowsky-, Nissl-, Holzer-, Mallory-, Heidenhain-, “fat-” and “pikrofuchsin”-stains. While a significant number of slides were no longer useable due to severe aging and bleaching, several slides were preserved quite well. The reader is invited to explore a selection of well-preserved slides here (see Figure 3 and slide-links). It is interesting to compare these examples to the original drawings drawn by van Gieson, provided in Bernard Sachs’s 1887 publication [35] on “amaurotic idiocy” (see Figure 4, and supplement 8).
Figure 3. Original historical slides from selected CNS sites of patient “Kn”. Specimen were well preserved stained Mallory, an additional neocortical specimen underwent some “fat-stain”, not otherwise specified (scanned with Zeiss AxioScan.Z1). The cerebral slides (lower row: neocortex and hippocampal region) illustrate the severe cortical pathology characterized by shrinking of the cortical band with profound loss of neurons. Residual neurons show the typical swollen, ballooned morphology of a CNS lipidosis. When going into detail on large magnifications, the neuronal pathology is seen well in brainstem also, esp. in the medulla/olive specimen (upper row, middle). Within the hippocampus proper there seems to be profound loss of pyramidal neurons, which could represent hippocampal sclerosis (this specimen was not available in other stains).
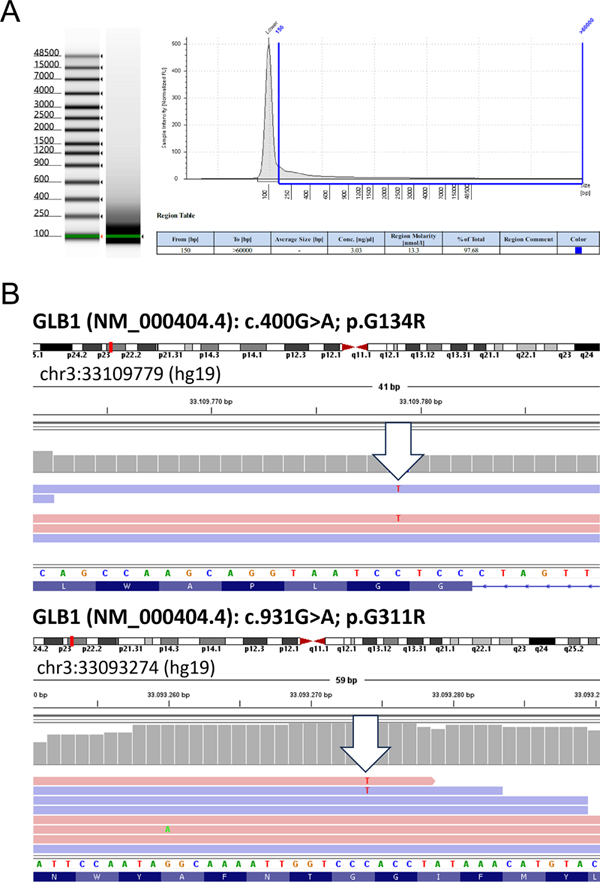
Figure 4. Original drawings by I. van Gieson (1866-1913), from Sachs’s publication 1887 [36] (Courtesy of the National Library of Medicine). See supplement 8. Have the original materials been used for modern molecular approaches and what was the result? To the best of our knowledge, the original materials of the patient Johann Knott presented here so far were not used for any modern molecular analysis later on, although materials participated in one subsequent ganglioside quantification analysis [15]. For this re-visitation, we performed a genetic analysis using a selected fraction of Kn’s archived material using only sections no longer usable for microscopy due to aging effects. This procedure was covered by a positive vote of the ethics commission at Munich University/LMU (Nr. 22-1021): Materials & Methods: Formalin-fixed paraffin-embedded (FFPE) cerebellar sections from the Historical Archive of the Max Planck Institute for Psychiatry in Munich were used as follows. Genomic DNA was extracted from three stained sections. The slides were incubated in xylene (Bergchemie J. C. Bröcking) for 14 days to enable lifting of the cover slip. Tissue was overlayed with Incubation Buffer containing Proteinase K (Maxwell FFPE Plus DNA Kit, Promega), scraped from the surface using a scalpel and resuspended for transfer into a 1.5 ml reaction tube to perform DNA extraction using the Maxwell FFPE Plus DNA Kit (Promega). DNA was quantified using the QuantiFluor ONE dsDNA System (Promega) and 20 ng were subjected to the Infinium HD FFPE Restore Protocol (Illumina, Zymo). Finally, 1 µl of the restored gDNA was inspected by Genomic DNA ScreenTape assay (Agilent) to determine DNA Integrity Number (DIN) and concentration (Figure 5A). Although DNA integrity was low (1.0), the Exome 2.0 Kit (Twist Bioscience) could be successfully applied for library preparation and sequencing was performed on a NovaSeq 6000 device. After base calling and adapter trimming, alignment to the human reference genome (Homo_sapiens.GRCh37.75_complete) and variant calling were performed using Dragen v07.021.624.3.10.10. Variants were annotated using the variant effect predictor (v100) for functional annotations, pathogenicity scores, population allele frequencies and to determine the effect of called variants on genes, transcripts, and protein sequence. Variants were manually inspected with the integrative genomics viewer (IGV). Results are illustrated in figure 5.
Figure 5. (A) DNA fragment analysis. Fragment analysis of gDNA isolated from formalin-fixed paraffin-embedded tissue shows high DNA degradation. The figure shows DNA fragment sizing and quantification using Genomic DNA ScreenTape assay. (B) IGV screenshots showing representative images of the two heterozygous mutations: GLB1 (NM_000404.4): c.400G>A; p.G134R (top) and GLB1 (NM_000404.4): c.931G>A; p.G311R (bottom). Both mutations have been described in patients with GM1 gangliosidosis [28]. Epilogue Amaurotic idiocy (AI), as exemplified by Tay-Sachs-Disease (TSD), had remained a fatal, enigmatic condition for a long period of time. AI’s impressive cellular pathology, affecting neuronal cells throughout the nervous system quite specifically, was recognized as early as 1887 ([36] supplement 8). After separating the tissue pathology later to be known as ceroid-lipofuscinosis [62; 46-48] classic neuromorphology for decades had not much to contribute to the differentiation of AI, for its CNS pathology appeared rather uniform [25]. Significant advances in this field were largely driven by biochemical insight into composition and distribution of central nervous compounds and by applying newly emerging techniques between 1920 and 1960, exemplified by the works of Klenk [19-21], Kuhn & Wiegandt [24], Svennerholm [53] and Sandhoff ([42], [45]). Sandhoff’s original observation [14] revisited here opened the door to the realization that TSD-like AI was not a homogenous entity, but a spectrum of neurometabolic diseases [2]. Triggered by detailed analysis of Johann Knott (Kn) and subsequent findings the field expanded and evolved rapidly ([2], [16], [44], [45]) leading to in depth characterization of the different forms of lipid storage disease, including enzymes & genes involved. Thus, GM1-gangliosidosis today can be summarized as a rare autosomally inherited lipid catabolism defect caused by deficiency of beta-galactosidase, encoded by the gene GLB1 on chromosome 3, leading to lysosomal storage of ganglioside GM1 and its sialic-free residue GA1 ([2], [16], [28]] encompassing various clinical forms ([3], [16], [52]). The term “AI” has long been abandoned and is now replaced by more specific terms representing our refined understanding. Acknowledgements
We thank: |
||||||||||||
|
Supplementary material |
||||||||||||
|
Slide links
|
||||||||||||
|
References 1. Annese J, Schenker-Ahmed NM, Bartsch H, Maechler P, Sheh C, Thomas N, Kayano J, Ghatan A, Bresler N, Frosch MP, Klaming R, Corkin S. Postmortem examination of patient H.M.'s brain based on histological sectioning and digital 3D reconstruction. Nat Commun. 2014; 5: 3122. https://doi.org/10.1038/ncomms4122 2. Breiden B, Sandhoff K. Lysosomal glycosphingolipid storage disease. Ann Rev. Biochem 2019; 88: 461 485. https://doi.org/10.1146/annurev-biochem-013118-111518 3. Caciotti A, Garman SC, Rivera Colon Y, Procopio E, Catarzi S, et al. 2011. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 1812:782–90. https://doi.org/10.1016/j.bbadis.2011.03.018 4 Czech H. Forschen ohne Skrupel. Die wissenschaftliche Verwertung von Opfern der Psychiatriemorde in Wien. In: Eberhard G, Neugebauer W; Von der Zwangssterilisierung zur E.rmordung. Zur Geschichte der NS Euthanasie in Wien Teil 2; 143 163; Wien/Köln/Weimar, Böhlau 2002. 5. Czech H, Weindling P, Druml C. From scientific exploitation to individual memorialization: Evolving attitudes towards research on Nazi victims' bodies. Bioethics 2021; 35 (6): 508 517. https://doi.org/10.1111/bioe.12860 6. Desnick RJ, Kaback MM (ed) Tay Sachs Disease, Academic Press, London Sand Diego 2001. 7. Diamond EF. Reflections on the 50th anniversary of the Nuremberg doctor’s trials. The Linacre Quarterly 1997; 64 (2): 17 20. https://doi.org/10.1080/20508549.1999.11878376 8. Escola J. [Über die Prozessausbreitung der amaurotischen Idiotie im Zentralnervensystem in verschiedenen Lebensaltern und Besonderheiten der Spätform gegenüber der Pigmentatrophie]. Arch.f. Psychiatrie u. Zeitschrift f. d. ges. Neurologie 1961; 202: 95 112. https://doi.org/10.1007/BF00342813 9. Escola Pico J. Über die Feinstruktur der Speichersubstanzen bei der infantilen Form der familiären amaurotischen idiotie. Acta Neuropathol 1964; 3: 289 294. https://doi.org/10.1007/BF00684405 10. Escola Pico J. Über die Ultrastruktur der Speichersubstanzen bei Spätfällen von familiärer amaurotischer Idiotie. Acta Neuropathol 1964; 3: 309 318. https://doi.org/10.1007/BF00691839 11. Gravel RA, Kaback MM, Proia RL, Sandhoff K et al. (2001) The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic & molecular bases of inherited disease, 8th edn. McGraw Hill, New York, pp 3827–3876. 12. Haymaker W, Neuburger KT, Hurteau WW (ed). Amaurotic family idiocy (slide 52). In: Atlas of Neuropathology prepared at the Army medical museum, Office of the surgeon general, U.S. Army, 1944. http://resource.nlm.nih.gov/41220590R 13. Hudson L. From small beginnings: the euthanasia of children with disabilities in Nazi Germany. J Pediatr Child Health 2011; 47: 508 511. https://doi.org/10.1111/j.1440-1754.2010.01977.x 14. Jatzkewitz H, Sandhoff K. On a biochemically special form of infantile amaurotic idiocy. Biochim. Biophys. Acta 1963; 70; 354 356. https://doi.org/10.1016/0006-3002(63)90764-9 15. Jatzkewitz H, Pilz H, Sandhoff K. The quantitative determination of gangliosides and their derivatives in different forms of amaurotic idiocy. J Neurochemistry 1965; 12: 135 144. https://doi.org/10.1111/j.1471-4159.1965.tb06749.x 16. Johnson WG. Galactosidase deficiency: GM1 gangliosidosis, Morqio B disease, and galactosialidosis. In: Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, Volume 1, 6th edition (ed. Rosenberg RN and Pascual JM), Academic Pres, London San Diego 2020 17. Kipfelsberger T. Die Konfrontation der Associationsanstalt Schönbrunn mit den nationalsozialistischen „Euthanasie“ Maßnahmen. Diss. med. TU München, 2017 18. Kinzelbach A, Neuner S, Hohendorf G, Buschmann M, Rauh P. Between institutional routine, hereditary health policy, and “NS euthanasia” killings. The dissection laboratory (prosector position) at the German Research Institute for Psychiatry (1926 1962). Medizinhist J 2022; 57: 332 362. https://doi.org/10.25162/mhj-2022-0012 19. Klenk E. Die Fettstoffe des Gehirns bei Amaurotischer Idiotie und Niemann Pick’scher Krankheit. Ber Ges Physiol 1937; 96: 659 660. 20. Klenk E. Niemann Pick’sche Krankheit und Amaurotische Idiotie. Hoppe Seyler’s Z Physiol Chem 1939; 262: 128 143. 21. Klenk E. Über die Ganglioside, eine neue Gruppe von zuckerhaltigen Gehirnlipoiden. Hoppe Seyler’s Z. Physiol. Chem. 1942; 273:76–86 22. Kolter T. Ganglioside biochemistry. International Scholarly Research Network. ISRN Biochemistry2012, Article ID 506160, 36 pages; https://doi.org/10.5402/2012/506160 23. Krivit W, Desnick RJ, Lee J, Moller F, Sweeley CC, Snyder PD, Sharp HL. Generalized accumulation of neutral glycoshpingolipids with GM2 ganglioside accumulation in the brain – Sandhoff’s disease (Variant of Tay Sachs Disease). Am J Med 1972; 52: 763 770. https://doi.org/10.1016/0002-9343(72)90082-4 24. Kuhn R, Wiegandt H. Die Konstitution der Ganglio N Tetraose und des Gangliosids G1. Chem. Ber. 1963; 96: 866 880. 25. Lake BD. Lysosomal enzyme deficiencies. In: Greenfield’s neuropathology, 4th edition, editors Adams JH, Corsellis JAN, Duchen LW. Edward Arnold, London 1984, 491 572. 26. Landing BH, Silverman FN, Craig JM, Jacoby MD, Lahey ME, Chadwick DL. Familial neurovisceral lipidosis. Am J Dis Child 1964; 108: 503 22. PMID: 14209687. 27. Lyon G, Kolodny EH, Pastores GM. Neurology of hereditary metabolic diseases of children, 3rd edition. Mc Graw Hill, New York 2006 28. Nicoli ER, Annunziata I, D’Azzo A, Platt FM, Tifft CJ, Stepien KM. GM1 Gangliosidosis – A Mini Review. Frontiers in Genetics 2021; 12. Article 734878. https://doi.org/10.3389/fgene.2021.734878 29. Norman RM, Urich H, Tingey AM, Goodbody RA. Tay Sachs disease with visceral involvement and its relation to Niemann Pick disease. J Pathol Bacteriol 1959; 78: 409 421. https://doi.org/10.1002/path.1700780208 30. O’Brien JS, Stern MB, Landing BH, O’Brien JK, Donnel GN. Generalized gangliosidosis: another inborn error of ganglioside metabolism? Am J Dis Child. 1965; 109: 338 346. PMID: 14261015. 31. Okada S, O’Brien JS. Generalized Gangliosidosis: Beta Galactosidase Deficiency. Science; 1968; 160: 1002 1004. https://doi.org/10.1126/science.160.3831.1002 32. Peiffer J. Assessing the neuropathological research carried out on victims of the “euthanasia” programme. Medizinhist J 1999; 34: 339 355. PMID: 10783589. 33. Pilz H, Müller D, Sandhoff K, ter Meulen V. Tay Sachssche Krankheit mit Hexosaminidase Defekt. Deutsch Med Wschr 1968; 39: 1833 1839 and 1843 1845. https://doi.org/10.1055/s-0028-1110836 34. Pilz H, Sandhoff K, Jatzkewitz H. A disorder of ganglioside metabolism with storage of ceramide lactoside, monosialo ceramide lactoside and tay sachs ganglioside in the brain. J Neurochemistry 1966; 13: 1273 1282. https://doi.org/10.1111/j.1471-4159.1966.tb04290.x 35. Sachs B. A family form of idiocy. Generally fatal and associated with early blindness (amaurotic family idiocy) New York Medical Journal, May 30, vol. 64 1896: 1 22. http://resource.nlm.nih.gov/101501959 36. Sachs B. On arrested cerebral development with special reference to its cortical pathology. J of Nervous and Mental disease, Vol XIV 1887 37. Sacrez R, Juif JG, Gigonnet JM, Gruner JE: La maladie de Landing, ou idiote amaurotique infantile precoce avec gangliosidose generalisee, Pediatrie 1967: 22: 143. PMID: 4973280. 38. Samuels S, Korey SR, Gonatas J, Terry RD, Weiss M. Studies in Tay Sachs disease. IV. Membranous cytoplasmic bodies. J Neuropath Exp Neurol 1963; 22:81–97. https://doi.org/10.1097/00005072-196301000-00005 39. Sandhoff K, Telephone conversations with BS Kasper on 22.3.2022 (2h), 27.9.2022 (1h), 23.12.2022 (1h) 40. Sandhoff K, Andreae U, Jatzkewitz H. Deficient hexosaminidase activity in an exceptional case of Tay Sachs disease with additional storage of kidney globoside in visceral organs. Life Sciences 1968; 7: 283 288. https://doi.org/10.1016/0024-3205(68)90024-6 41. Sandhoff K. Variation of beta N acetylhexosaminidase pattern in Tay Sachs Disease. FEBS letters 1969; 4 (4): 351 354. https://doi.org/10.1016/0014-5793(69)80274-7 42. Sandhoff K, Harzer K. Gangliosides and Gangliosidoses: Principles of molecular and metabolic pathogenesis. J Neurosci 2013; 33 (25): 10195 10208. https://doi.org/10.1523/JNEUROSCI.0822-13.2013 43. Sandhoff K. Die amaurotische Idiotie des Menschen als Störung des Glycosphingolipoidstoffwechsels. Thesis, Munich University (1965) DISSERTATION [German] 44. Sandhoff K. The GM2 Gangliosidoses and the Elucidation of the beta Hexosaminidase System. Advances in Genetics 2001; 44: 67 91. https://doi.org/10.1016/s0065 2660(01)44072 7 45. Sandhoff K. My journey into the world of sphingolipids and sphingolipidoses. Proc Jpn Acad Ser B Phys Biol Sci 2012; 88: 554 582. https://doi.org/10.2183/pjab.88.554 46. Spielmeyer W. Über familiäre amaurotische Idiotien. Neurol. Centralblatt 1905; 24: 620 621 47. Spielmeyer W. Über eine besondere Form von familiärer amaurotischer Idiotie. Neurol Centralblatt 1906; 25: 51 55 48. Spielmeyer W. Klinische und anatomische Untersuchungen über eine besondere Form von familiärer amaurotischer Idiotie. Histol und Histopathol 1908 (2): 193 251 & Tafeln XXII XXIII 49. Seitelberger F, Vogel G, Stepan H. Spätinfantile amaurotische Idiotie. Archiv für Psychiatrie und Zeitschrift f. d. ges. Neurologie 1957; 196: 54 190. https://doi.org/10.1007/bf00354507 50. Simonati A, Williams RE. Neuronal ceroid lipofuscinosis: The multifaceted approach to the clinical Issues, an overview. Front Neurol. 2022 Mar 11;13: 811686. https://doi.org/10.3389/fneur.2022.811686 51. Suzuki K, Chen GC: Brain ceramide hexosides in Tay Sachs disease and generalized gangliosidosis (GM1 gangliosidosis), J. Lipid Res. 8: 105 113, 1967. PMID: 14564716. 52. Suzuki Y, Oshima A, Nanba E. 2001. β Galactosisdase deficiency (β galactosidosis): GM1 gangliosidosis and Morquio B disease. In: The Metabolic and Molecular Bases of Inherited Diseases, ed. CR Scriver, AL Beaudet, WS Sly, D Valle, pp. 3775 3809. New York: McGraw Hill 53. Svennerholm L. The chemical structure of normal human brain and Tay Sachs gangliosides. Biochem. Biophys Research Communications 1962; 9 (5): 436 441. https://doi.org/10.1016/0006-291x(62)90030-x 54. Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Transactions of the Ophthalmological Society U.K 1881; 1: 55–57. https://doi.org/10.1001/archneur.1969.00480070114014 55. Terry RD and Korey SR Membranous cytoplasmic granules in infantile amaurotic idiocy. Nature 1960; 188, 1000 1002. https://doi.org/10.1038/1881000a0 56. Terry RD, Weiss M. Studies in Tay Sachs disease II. Ultrastructure of the cerebrum. J Neuropathol Exp Neurol 1963; 22: 18 55. https://doi.org/10.1097/00005072-196301000-00003 57. Vogt H. Über familiäre amaurotische Idiotie und verwandte Krankheitsbilder. Monatsschrift für Psychiatrie und Neurologie 1905; 18: 161 171, 310 357. 58. Walkley SU. Cellular pathology of lysosomal storage disease. Brain Pathology 1998; 8: 175 193. https://doi.org/10.1111/j.1750-3639.1998.tb00144.x 59. Weindling P, Hohendorf G, Hüntelmann AC, Kindel J, Kinzelbach A, Loewenau A, Neuner S, Palacz MA, Zingler M, Czech H. The problematic legacy of victim specimens from the Nazi era: Identifying the persons behind the specimens at the Max Planck Institutes for Brain Research and of Psychiatry. J Hist Neurosci. 2021 Oct 18:1 22. https://doi.org/10.1080/0964704x.2021.1959185 60. Zeidman LA. Hans Jacob and brain research on Hamburg “euthanasia” victims “Awaiting further brains!”. Neurology 2017 ;88: 1089–1094. https://doi.org/10.1212/wnl.0000000000003712 61. Zeidman LA. Brain Science Under The Swastika. Ethical violations, resistance, and victimization of neuroscientists in Nazi Europe. Oxford University press, Oxford 2020. 62. Zeman W, Dyken P. Neuronal ceroid lipofuscinosis (Batten’s disease): relationship to amaurotic family idiocy? Pediatrics 1969; 44: 570-583. PMID: 5346636.
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |