|
|
|
Free Neuropathology 4:2 (2023) |
|
Review |
|
Neuromuscular disease: 2023 update |
|
Marta Margeta |
|
Department of Pathology, University of California, San Francisco, CA, USA |
|
Corresponding author: |
|
Submitted: 07 February 2023 |
|
Keywords: Long COVID, Guillain-Barré syndrome, Wallerian degeneration, Nodes of Ranvier, Muscle regeneration, Chaperonopathy, Target fiber, Inclusion body myositis, Facioscapulohumeral dystrophy, Myotonic dystrophy, Amyotrophic lateral sclerosis |
|
Abstract This review highlights ten important advances in the neuromuscular disease field that were reported in 2022. As with prior updates in this article series, the overarching topics include (i) advances in understanding of fundamental neuromuscular biology; (ii) new / emerging diseases; (iii) advances in understanding of disease etiology and pathogenesis; (iv) diagnostic advances; and (v) therapeutic advances. Within this general framework, the individual disease entities that are discussed in more detail include neuromuscular complications of COVID-19 (another look at the topic first covered in the 2021 and 2022 reviews), DNAJB4-associated myopathy, NMNAT2-deficient hereditary axonal neuropathy, Guillain-Barré syndrome, sporadic inclusion body myositis, and amyotrophic lateral sclerosis. In addition, the review highlights a few other advances (including new insights into mechanisms of fiber maturation during muscle regeneration and fiber rebuilding following reinnervation, improved genetic testing methods for facioscapulohumeral and myotonic muscular dystrophies, and the use of SARM1 inhibitors to block Wallerian degeneration) that will be of significant interest for clinicians and researchers who specialize in neuromuscular disease. Abbreviations AD - autosomal dominant, ADPR - adenosine diphosphate ribose, AIDP - acute inflammatory demyelinating polyneuropathy, ALS - amyotrophic lateral sclerosis, AMAN - acute motor axonal neuropathy, AMSAN - acute multifocal sensorimotor neuropathy, AR - autosomal recessive, CNM - centronuclear myopathy, COX - cytochrome c oxidase, COVID-19 - Coronavirus Disease 2019, DM - dystrophia myotonica (myotonic dystrophy), EMG - electromyography, FSHD - facioscapulohumeral dystrophy, GBS - Guillain-Barré syndrome, IEFND - intraep-idermal nerve fiber density, IVIg - intravenous immunoglobulin, LMN - lower motor neuron, MAC - membrane attack complex, Mfn2 - mitofusin 2, MHC-I - major histocompatibility complex I, MND - motor neuron disease, MyHC - myosin heavy chain, NCS - nerve conduction studies, NoR - node(s) of Ranvier, PASC - post-acute sequalae of SARS-CoV-2 infection, PM-Mito - polymyositis with mitochondrial abnormalities, SC - Schwann cell, SFN - small fiber neuropathy, sIBM - sporadic inclusion body myositis, STR - short tandem repeat, XLMTM - X-linked myotubular myopathy In this annual update, I will briefly describe ten neuromuscular field advances from 2022 that I consider to be most important and/or interesting; as in the prior updates (Margeta, 2020, 2021, 2022), these advances will be grouped into different “discovery clusters” and listed in no particular order. Advances in fundamental neuromuscular biology with implications for neuromuscular disease 1. Regulation of the neonatal-adult maturation checkpoint during skeletal muscle regeneration In the past two reviews (Margeta, 2021, 2022), I described the crosstalk between macrophages and satellite cells (muscle stem cells) that initiates skeletal muscle repair. In this review, the focus will be on a later stage of myofiber regeneration, which recapitulates muscle development and involves myofiber growth and maturation from a fetal to the adult state. In mammals, there are seven distinct isoforms of myosin heavy chain (MyHC; the major contractile component of thick filaments) – two isoforms that are normally expressed only during development [embryonic (MyHC-emb / MHC3) and neonatal (MyHC-neo / MHC8)] and four isoforms that are expressed in mature muscle fibers [one slow isoform (MHC-slow / MHC7; type 1 fibers) and three fast isoforms: MyHC-2A / MHC2 (type 2A fibers), MyHC-2X / MHC1 (type 2X fibers) and MyHC-2B / MHC4 (type 2B fibers)] (Schiaffino et al., 2015). During muscle repair, regenerated myofibers mature over ~14 days by recapitulating developmental sequence of MyHC expression: newly formed fibers first express MyHC-emb, followed by MyHC-neo, and finally one of the adult MyHC isoforms. [This aspect of the regeneration process is diagnostically useful: in adult muscle, immunohistochemistry for developmental MyHC isoforms can be used to detect regenerating fibers [(Sewry et al., 2021) and Fig. 1.].
Figure 1. Regenerating muscle fibers express developmental myosin heavy chain isoforms, MyHC-emb (MHC3) and/or MyHC-neo (MHC8). Dual immunohistochemical stain for MyHC-emb (red) and MyHC-neo (brown), here performed on cryosections, can be used to detect regenerating fibers at different stages of maturation. (A) In normal adult skeletal muscle, there is no staining for either MyHC-emb or MyHC-neo. (B) A case of immune-mediated necrotizing myopathy (IMNM) shows frequent regenerating fibers, most of which are in the early stages of maturation and express only MyHC-emb (an example is highlighted by the green arrow). A few regenerating fibers are in the late stages of maturation and express only MyHC-neo (an example is highlighted by the orange arrow), while several regenerating fibers are transitioning from an early to a late maturation stage and co-express both MyHC-emb and MyHC-neo (dark orange sarcoplasmic staining; an example is highlighted by the black arrow). (C) A case of sporadic inclusion body myositis (sIBM) shows abundant regenerating fibers spanning the entire spectrum of maturation. (D) In dermatomyositis (DM), both early and late regenerating fibers are mainly found in the perifascicular areas. Scale bars: A and D, 200 µm; B and C, 50 µm. Wang and co-authors have now elucidated molecular and cellular mechanisms that regulate the neonatal / adult transition checkpoint, a key step in this maturation sequence (Wang et al., 2022). Using a myotoxin-induced model of skeletal muscle injury, they showed that expression of mitofusin 2 (Mfn2; a mitochondrial membrane protein that initiates mitochondrial fusion and is induced in activated satellite cells) is required for maturation of newly formed myofibers: in Mfn2-deficient mice, myotoxic muscle injury led to formation of early (centrally nucleated, MHC3-expressing) regenerating fibers, but these fibers were smaller than regenerating myofibers in wild-type (wt) mice and ultimately arrested their maturation at the neonatal (MHC8-expressing) developmental stage. This maturation defect was not due to a metabolic abnormality in satellite cells; rather, Mfn2 deletion led to nuclear translocation of transcription factor NFATC2 (nuclear factor of activated T cells 2) and increased expression of HIF1α (hypoxia-induced factor 1α), ultimately resulting in the lack of repressive methylation marks at the MHC8 genetic locus, which are required for the neonatal / adult fate transition. In wt animals, expression of HIF1α protein was induced following muscle injury, but its level returned to the preinjury baseline within 5 days; in Mfn2-null animals, HIF1α levels remained elevated 14 days post-injury. Importantly, sustained HIF1α signaling was both necessary and sufficient for the arrest of regenerating myofibers at the neonatal stage: treatment of Mfn2-/- mice with PX-478 (a chemical compound that reduces HIF1α levels) for 14 days, starting at 14 days after muscle injury, enabled Mfn2-/- regenerating myofibers to escape the maturation arrest, adopt the adult fate, and grow closer to a normal adult fiber size. The same rescue could be achieved through genetic deletion of HIF1α or through inhibition of H3K27 demethylases; conversely, the effect of Mfn2 deletion could be replicated by genetic manipulation of VHL (von-Hippel Lindau tumor suppressor) signaling, which leads to HIF1α stabilization independent of Mfn2 signaling. Finally, the authors used an ischemic model of muscle injury to show that in wt mice, repressive methylation marks at the MHC8 locus started appearing on day 9 (coincidental with reperfusion), followed by fiber growth and transition to the adult fate on day 10, and complete downregulation of MHC8 expression by day 12. In contrast, myofiber maturation was significantly accelerated in animals lacking HIF1α, with completely differentiated fibers lacking any MHC8 expression present on day 8. Taken together, these data indicate that Mfn2/HIF1α signaling regulates myofiber fate specification through the epigenetic control of MyHC-neo / MHC8 expression, and that this checkpoint enables synchronous development of all skeletal muscle tissue elements. However, it should be noted that all experiments in this study were done on the mouse tibialis anterior muscle, which is largely composed of type 2 fibers; thus, it remains to be shown whether a similar maturation checkpoint also affects type 1 fibers. What is the relevance of this regulatory pathway for human disease? While that question needs to be investigated in much greater detail in the future, initial experiments performed by Wang and co-authors offer some tantalizing clues. Because maturation-arrested regenerating fibers are reminiscent of atrophic, centrally nucleated fibers seen in severe centronuclear myopathies (CNMs), they evaluated biopsies from infantile patients with CNM 1 (caused by mutations in dynamin 2) and CNM 2 / X-linked myotubular myopathy (XLMTM, caused by mutations in myotubularin 1); in both diseases, they detected myofibers strongly positive for MHC8 as well as nuclear NFATC2 and sarcoplasmic carbonic anhydrase 3 (a HIF1α target). While MHC3+ and MHC8+ positive fibers can be seen in any myopathy that involves significant fiber necrosis (Fig. 1), they are generally not very numerous in congenital myopathies, raising the possibility that the Mfn2/HIF1α-controlled maturation checkpoint is specifically dysregulated in CNMs. Interestingly, a different 2022 study demonstrated that epigenetic alterations are a conserved feature of XLMTM and that histone deacetylase inhibition is a promising therapeutic strategy for this severe muscle disease (Volpatti et al., 2022). While altered muscle development is one of the epigenetically modified pathways uncovered by Volpatti et al., it remains to be shown whether changes in the methylation of the MHC8 genetic locus are part of the XLMTM epigenetic disease signature. Interestingly, autosomal dominant mutations in Mfn2 cause axonal Charcot-Marie-Tooth disease type 2A (CMT 2A) but are not known to produce a significant myopathic phenotype. It is possible that CMT 2A patients exhibit atypical muscle development and/or aberrant response to muscle injury that contributes to their weakness but has been masked by their peripheral neuropathy; that needs to be explored by future studies. In addition, it needs to be investigated whether pharmaceutical treatments that turn off this maturation checkpoint can accelerate muscle repair following injury and/or prove beneficial for any muscle disease characterized by significant fiber necrosis and regeneration. Newly defined / emerging neuromuscular diseases 2. Neuromuscular complications of COVID-19: Post-Acute Sequelae of SARS-CoV-2 Infection (“Long COVID”) Syndrome COVID-19, the novel infectious disease caused by SARS-CoV-2, primarily targets the respiratory system but also affects many other tissues and organs, including the PNS. In the past two reviews (Margeta, 2021, 2022), I discussed the emerging understanding of acute neuromuscular complications of COVID-19; in this review, I will summarize what is currently known about neuromuscular involvement in the post-acute sequelae of SARS-CoV-2 infection (PASC) syndrome (also known as “post-COVID conditions” or “long COVID”). The CDC (Centers for Disease Control and Prevention, a branch of the United States government) defines PASC as continuation of COVID-19 symptoms – or the emergence of new symptoms – four weeks or longer after initial SARS-CoV-2 infection; given this broad definition and the continuously changing spectrum of viral variants, the true prevalence of PASC is not known, but the current estimates range from 5-30%. While PASC can include a wide array of symptoms, some of the most common and particularly debilitating concerns (including fatigue / exertional intolerance, dysautonomia, and sensory symptoms such as paresthesias and numbness) suggest that the PNS is affected. Two small retrospective studies published in 2022 explored whether autonomic and sensory symptoms experienced by PASC patients can be attributed to small fiber neuropathy (SFN). The first study included 13 patients (8 women and 5 men) who had acute COVID-19 infection in March-April 2020 and developed paresthesias 0-2 months later; the patients were evaluated by electromyography / nerve conduction studies (EMG/NCS) and two punch skin biopsies (from distal leg and proximal thigh) to determine intraepidermal nerve fiber density (IEFND) (Abrams et al., 2022). Of these 13 patients, only one had severe COVID, and none had a history of prior neurologic symptoms or evidence of large fiber polyneuropathy on EMG/NCS. Seven of 13 patients also reported orthostatic discomfort; four of these seven patients underwent autonomic function testing, with abnormal findings reported in three. Reduced IEFND was seen in 46% patients (6 of 13; all female), all of whom had clinical findings consistent with SFN (mainly a decreased pinprick sensation in distal legs); the biopsy findings were also more severe distally. In contrast, most patients with normal biopsy results showed no objective sensory abnormalities on neurologic exam. The second study enrolled 17 patients (11 women and 6 men) who developed COVID-19 between February 2020 and January 2021 and presented with new neuropathy symptoms 0.1 to 14.9 months following COVID onset; again, only 1 of these 17 patients had a severe COVID infection (Oaklander et al., 2022). In this cohort, 16.7% of electrodiagnostic studies showed abnormal findings, while 62.5% of lower leg skin biopsies and 50% of upper thigh biopsies showed reduced IEFND. Importantly, in both studies the clinicopathologic evaluation was performed with a significant delay due to pandemic-related disruptions in health care delivery; thus, it is possible that partial or complete small fiber recovery occurred in some patients in the interval between the symptom onset and assessment, leading to false-negative test results. Both studies should be considered preliminary given their small size and confounding by referral bias; however, the reported findings are very similar and – if conformed in larger cohorts – suggest that the PASC-associated peripheral neuropathy reflects small nerve fiber injury. The underlying disease mechanisms also remain to be determined; however, a delay between COVID infection and neurologic symptom onset in all but one of these 30 pa-tients, along with therapeutic effectiveness of the repeated IVIg treatment in 5 patients who received it, suggest that the PASC-associated SFN is due to post-infectious dysregulation of the immune system. Persistent fatigue is experienced by 50-70% of PASC patients, making it one of the most common PASC symptoms. However, it has been challenging to determine whether this relatively subjective and nonspecific clinical symptom is caused by functional response of muscle tissue to circulating inflammatory mediators, microcirculatory abnormalities, or an underlying skeletal myopathy. To start addressing this question, Hejbøl and colleagues evaluated 16 patients (13 women and 3 men) with chronic post-COVID fatigue; none of the patients had been severely ill with COVID or hospitalized in intensive care unit, and clinicopathologic evaluation for myopathy (which included quantitative EMG and skeletal muscle biopsy) occurred 5-14 months following acute infection (Hejbøl et al., 2022). Among these 16 patients with PASC fatigue, 69% also had paresthesias, but none had abnormal deep tendon reflexes or altered vibratory sensation, and NCS were normal in all 16. In addition to the fatigue, 81% of study patients had myalgia, 50% objective muscle weakness, and 75% myopathic findings on EMG; serologic studies were generally unremarkable, except for one patient with a marginally elevated creatine kinase level and another with elevated TIF1-γ antibody titer but normal creatine kinase level. Remarkably, muscle biopsies from all 16 patients showed histologic and/or ultrastructural muscle fiber pathology; the findings were variable, but included prominent nucleoli and non-selective fiber atrophy (suggestive of fiber regeneration), basal lamina reduplication, myofibrillar disorganization, and mitochondrial abnormalities (Fig. 2). In addition, inflammation and/or MHC-I upregulation was present in 62% of biopsies, while capillary abnormalities (including basal lamina reduplication and capillary loss/degeneration) were detected in 75% of biopsies. Finally, one patient showed a loss of non-myelinated, small caliber axons in the intramuscular nerve twigs. The spectrum of findings seen in these specimens is very broad and does not point to a specific disease mechanism; however, both mitochondrial and capillary pathology could provide an explanation for fatigue, while basal lamina reduplication could be due to increased TGFβ (transforming growth factor β) levels, which often accompany chronic low-grade inflammation. Like the two SFN studies described above, this study has several important limitations, including small cohort size, lack of patients from later stages of the pandemic (who were infected by SARS-CoV-2 variants other than the ancestral strain), and lack of a control group (i.e., patients with COVID-19 who did not develop fatigue or other PASC symptoms); nonetheless, the findings suggest that muscle biopsy is the most sensitive test for diagnosis of the PASC-associated myopathy.
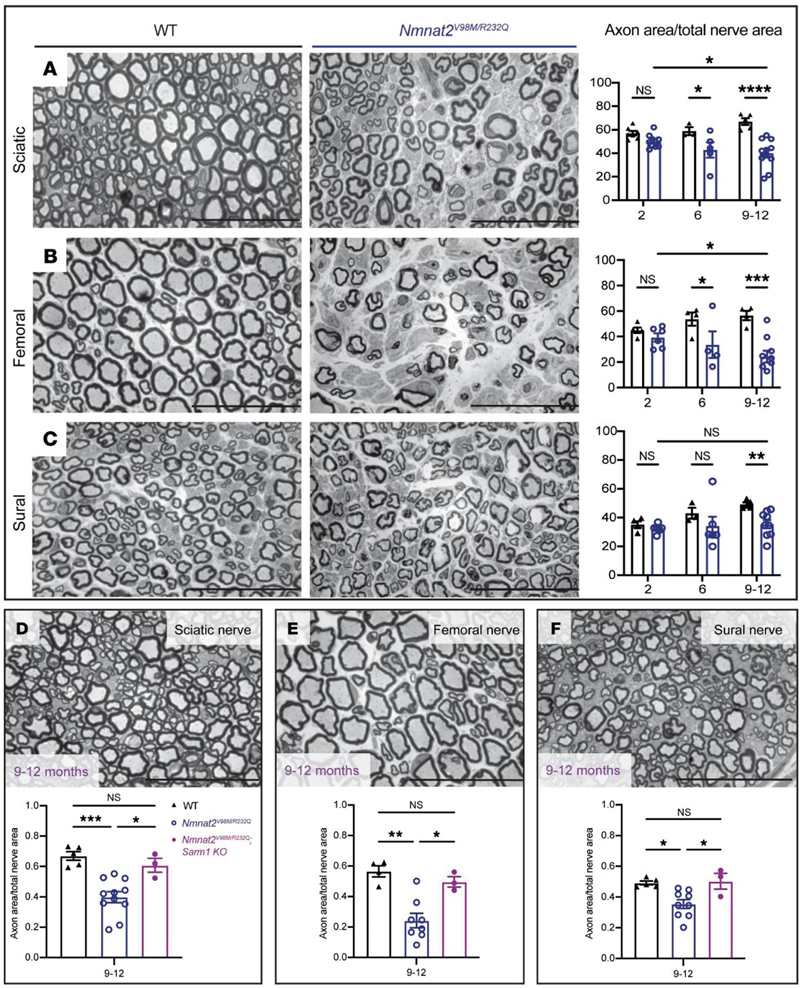
Figure 2. Myopathy as a cause of fatigue in long-term post-COVID-19 symptoms: Histopathological changes in muscle fibers. (a, b) Muscle fiber atrophy. (a) Atrophic fiber and (b) folds in the basal lamina, both in Patient 9. (c–f) Muscle fiber damage indicated by basal lamina proliferation. (c) Basal lamina duplication (arrow) in Patient 16. (d, e) Multiple myofiber basal lamina (d) and (e) higher magnification showing continuity between the different layers (arrows) in Patient 14. (f) From Patient 6, showing aggregation of nuclei, and multiple layers of myofiber basal lamina (arrow). (g–i) Myofibrillary disorganization: (g) streaming of Z-bands in Patient 14, (h) disorganized myofibrils in Patient 13, and (i) myofibril disorganization and a cytoplasmic body in Patient 1. (j–l) Mitochondrial changes: (J) a cytochrome c oxidase-negative fiber (blue) and (k) subsarcolemmal accumulation of structurally abnormal mitochondria from Patient 8, and (l) subsarcolemmal accumulation of ultrastructural normal-appearing mitochondria in Patient 9. Scale bars: k = 500 nm; c, e, h, and l = 1 μm; a, b, d, f, g, and i = 2 μm; j = 100 μm. [This figure and its legend are reproduced without modifications from (Hejbøl et al., 2022); this use is permitted under the Creative Commons Attribution 4.0 CC-BY-NC-ND International License.] Collectively, these three studies provide initial evidence that neuromuscular symptoms commonly seen in PASC patients are associated with – and likely attributable to – structural abnormalities of peripheral nerves and skeletal muscles; however, these preliminary findings should be replicated in larger and temporally more homogenous cohorts of PASC patients. Furthermore, additional work is required to establish whether similar nerve and muscle abnormalities are detectable in patients with myalgic encephalomyelitis / chronic fatigue syndrome, which shares many clinical features with the PASC syndrome. 3. DNAJB4-associated myopathy: A new chaperonopathy Molecular chaperones play a key role in the maintenance of protein homeostasis by facilitating folding of newly synthetized proteins and degradation of misfolded or damaged / unfolded proteins; the latter role is particularly critical in skeletal muscle, where mechanical stress leads to constant damage to sarcomeric proteins and where misfolded protein aggregates cannot be diluted through cell division. As a result, although chaperones and co-chaperones are ubiquitously expressed proteins, skeletal myopathy is generally the main (and often only) clinical manifestation of many chaperonopathies. DNAJ/HSP40 co-chaperones determine the specificity of the chaperone/client interaction and are highly evolutionarily conserved from yeast to humans; mutations in several DNAJ co-chaperones from the B family (DNAJB2, DNAJB5 and DNAJB6) cause neuromuscular disease, but prior to 2022 no human disease has been linked to DNAJB4. DNAJB6 is a close homolog of DNAJB4; it localizes to the Z-disc and, together with HSPB8 and BAG3, plays a critical role in the Z-disc maintenance via chaperone-assisted selective autophagy. Autosomal dominant mutations in DNAJB6 lead to two distinct clinical phenotypes, limb-girdle muscular dystrophy D1 and distal myopathy, both of which are histologically characterized by myofibrillar disorganization, protein aggregates, and rimmed vacuoles (Sarparanta et al., 2020). Last year, two separate research groups showed that mutations in DNAJB4 cause a similar but distinct skeletal myopathy that is inherited in either autosomal recessive (AR; Weihl et al., 2023) or autosomal dominant (AD; Inoue et al., 2023) manner. Weihl and co-authors used a reverse genetic approach to identify four patients from three different families who carry autosomal recessive, loss-of-function mutations in DNAJB4 that either severely abrogate protein expression (c.856A>T; p.Lys286Ter and c.785T>C; p.Leu262Ser) or lead to expression of a non-functional protein (c.74G>A; p.Arg25Gln). Clinically, these patients presented in the first decades of life with spinal rigidity and severe diaphragmatic weakness that led to early-onset respiratory failure. In contrast, Inoue and colleagues used a more traditional forward genetic approach: they identified a single family with an autosomal dominant, slowly progressive myopathy characterized by onset in the 3rd to 5th decade, asymmetric thumb and grip weakness, symmetric distal weakness, respiratory failure, scoliosis, and ultimately loss of ambulation, and showed that a heterozygous missense variant of DNAJB4 (c.270T>A; p.Phe90Leu) segregated with this clinical phenotype. Muscle pathology was similar in both AD and AR forms of DNAJB4 disease, with fiber size variation, rubbed out fiber centers on oxidative stains, rimmed vacuoles in atrophic fibers, and very large central inclusions / protein aggregates (up to 30-40 sarcomeres in length) in non-atrophic fibers that stained red on modified Gomori trichrome, had an amorphous “woolly” or granulofilamentous appearance on EM, and were immunopositive positive for p62, Hsp70, desmin, myotilin, filamin C, and (in the AD form of disease) for DNAJB4. Ultrastructurally, there was also widespread degeneration of sarcomeres, but Z-discs were relatively spared, with no definite Z-band widening or streaming. Interestingly, Inoue and co-authors showed that this pathology is more prominent in type 1 muscle fibers, providing explanation for the preferential clinical involvement of muscles that are type 1 fiber-predominant (soleus, diaphragm, paraspinals, as well as thenar and hypothenar muscles). Impressively, both groups generated model mice that recapitulated key aspects of human DNAJB4 disease, including its unique myopathologic features and preferential involvement of type 1 muscle fibers; this was true both for knockout Dnajb4-/- mice (which model AR human disease; created independently by both groups) and for Dnajb4F90L/+ knock-in mice (which model AD human disease; created by Inoue et al.), suggesting that the F90L DNAJB4 mutation has a dominant negative effect. Of course, a lot remains to be discovered about this new disease. For example, it remains to be established why type 1 muscle fibers are more vulnerable to DNAJB4 deficiency than type 2 fibers. In addition, it is not entirely clear why there are significant clinicopathologic differences between DNAJB6 and DNAJB4 diseases given the large degree of structural homology between these two proteins. One clue that has emerged from the work of Inoue et al. is a subtle difference in the subcellular localization of these two proteins: while DNAJB4 is localized to the close vicinity of the Z-disc, DNAJB6 directly colocalizes with this structure. Future studies will be necessary to delineate the functional differences between these two co-chaperones, particularly with respect to their roles in chaperone-assisted selective autophagy, and in formation and/or degradation of RNA stress granules. 4. NMNAT2-deficient, SARM1-dependent hereditary axonal neuropathy (“SARMopathy”) Wallerian degeneration is a highly regulated, active process of axon destruction; it is typically seen following acute mechanical or ischemic injury, but can also be caused by toxins, nutritional deficiencies, immune-mediated injury (further discussed in advance #5), and genetic alterations. Regardless of the underlying etiology, Wallerian degeneration is triggered by NAD+ hydrolase SARM1 (sterile alpha and Toll/interleukin-1 receptor motif-containing 1), the activity of which is regulated by the axonal NMN/NAD+ ratio (Osterloh et al., 2012; Figley et al., 2021). Under normal conditions, the activity of SARM1 (which is often described as “the central executioner of a cell-autonomous axon degeneration program”) is kept in check by a highly labile NAD+-generating enzyme NMNAT2 (nicotinamide mononucleotide adenylyltransferase 2); NMNAT2 is trafficked from the soma into the axon, where it catalyzes synthesis of NAD+ using NMN and ATP as substrates (Conforti et al., 2014). Axonal injury disrupts axon transport of NMNAT2, resulting in the buildup of NMN and depletion of NAD+ distal to the injury site; this increases the NMN/NAD+ ratio and activates SARM1, further depleting NAD+ levels and causing metabolic crisis that ultimately leads to axon fragmentation. Not surprisingly, deletion of NMNAT2 in mouse models leads to perinatal lethality; however, mice lacking both NMNAT2 and SARM1 are viable and protected against axonal degeneration. In humans, NMNAT2 deficiency is very rare; however, homozygous R232Q NMNAT2 variant (which causes severe reduction in NMNAT2 activity) has been linked to fetal akinesia deformation sequence (Lukacs et al., 2019), while homozygous T94M NMNAT2 variant (which reduces NMNAT2 thermal stability) causes mild sensorimotor axonal polyneuropathy with erythromelalgia (Huppke et al., 2019). (Erythromelalgia is a syndrome that consists of sporadic attacks of burning pain, redness, and swelling, typically affecting the lower extremities; it is most often caused by gain-of-function mutations in SCN9A, which encodes sodium channel Nav1.7.) In their tour-de-force study published last year, Dingwall and coauthors expanded the spectrum of human NMNAT2-associated disease by showing that heterozygous NMNAT2 loss-of-function mutations cause severe hereditary axonal neuropathy through a SARM1-dependent, macrophage-mediated non-cell autonomous pathway; consequently, the authors have termed this new disease “SARMopathy” (Dingwall et al., 2022). The two affected brothers described by Dingwall et al. share a very unique clinical phenotype that includes (i) repeated episodes of acute multifocal sensorimotor axonal neuropathy (AMSAN) that mimic Guillain-Barré syndrome (GBS), are typically triggered by acute infection, and include severe pain, erythromelalgia, flaccid quadriparesis, and respiratory failure; and (ii) chronic axonal motor neuropathy that progresses between these acute episodes and leads to distal weakness, scoliosis, and wheelchair dependence in the third decade of life. The patients carry compound heterozygous NMNAT2 variants (the previously identified pathogenic variant c.695G>A; p.Arg232Gln / R232Q and a novel variant c.292G>A; p.Val98Met / V98M); each of these variants was inherited from one of the patients’ asymptomatic parents and was shown to markedly reduce NMNAT2 enzymatic activity. To further study this new hereditary neuropathy, the authors generated Nmnat2V98M/R232Q model mice. Like the two patients, these model mice developed an age-dependent, motor-predominant axonal polyneuropathy that led to severe hindlimb wasting by 9-12 months of age and was characterized by a severe loss of axons in mixed and motor nerves (Fig. 3A-B); in contrast, sensory nerves were largely spared (Fig. 3C), as were motor neuron cell bodies in the ventral horn of the spinal cord. Unlike the two patients, the model mice did not develop AMSAN episodes or hypersensitivity to pain, possibly because they were housed in a pathogen-free animal facility and thereby spared from acute infections. As expected, the axonal neuropathy phenotype in model mice was rescued by simultaneous deletion of SARM1 (Fig. 3D-F), demonstrating that SARM1 activation was necessary for axon degeneration; a similar, but slightly less effective rescue was achieved by transgenic expression of a dominant negative variant of SARM1. More surprisingly, the SARM1 effect in this mouse model was not entirely cell-autonomous; instead, it was at least in part mediated by macrophage activation, and could therefore be blocked by macrophage depletion (via monoclonal antibody against colony stimulating factor 1 receptor). Importantly, macrophage depletion was effective not only when treatment was started at birth, but also when it was started at 4 months of age (by which point mice already developed significant neuropathy); this suggests that blocking ongoing axonal degeneration can tip the balance toward axonal regeneration and functional recovery.
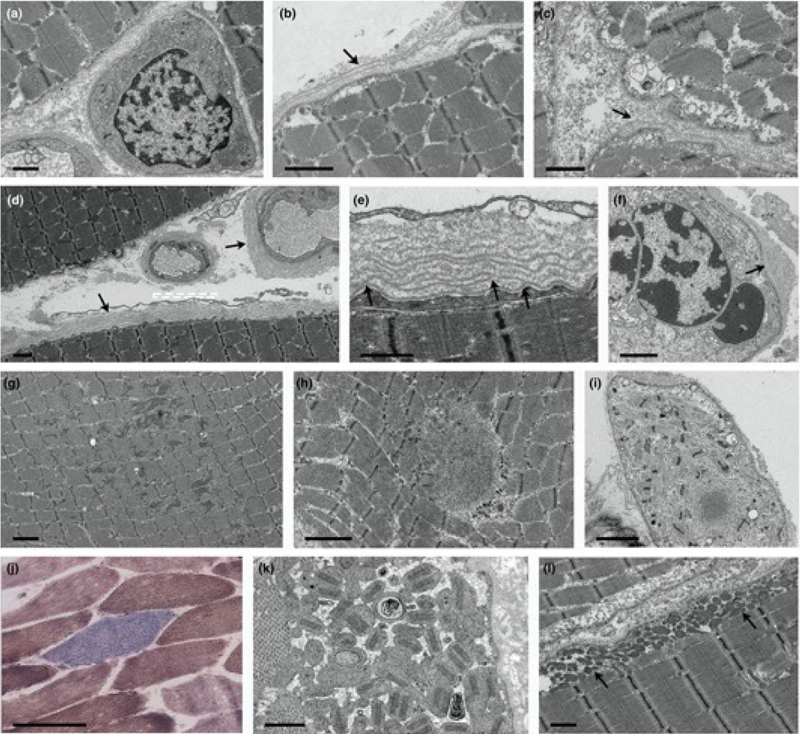
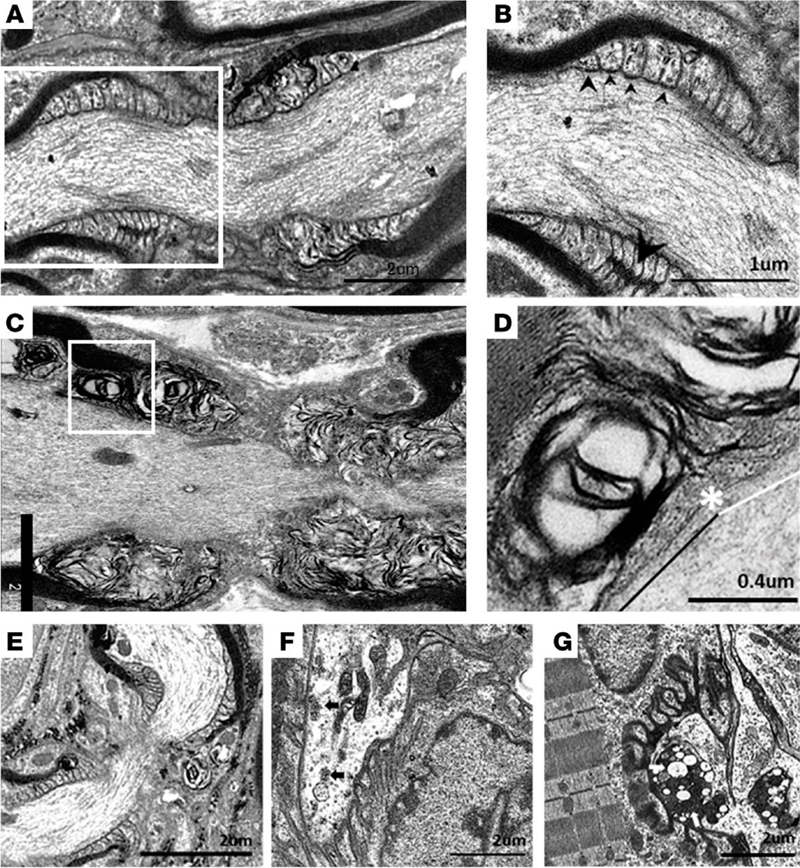
Figure 3. Nmnat2 variants cause progressive axon loss in mice in a SARM1-dependent manner. (A–C) Representative images of sciatic (A), femoral (B), and sural (C) nerves in 9–12-month-old Nmnat2V98M/R232Q (n = 9) or WT (n = 5) mice. Percent axonal area/total nerve area are indicated to the right (n = 4–11 mice per age cohort, per genotype). (D–F) Representative images of sciatic (D), femoral (E), and sural (F) nerves in 9–12-month-old Nmnat2V98M/R232Q; Sarm1-KO mice. Percent axonal area/total nerve area is calculated below each corresponding nerve (n = 3–11 mice per age cohort, per genotype). Statistical significance was determined by 2-way ANOVA with multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, 50 μm. [This figure and its legend were adopted from Figures 3 (A-C) and 5 (D-F) in (Dingwall et al., 2022); this use is permitted under the Creative Commons Attribution 4.0 CC-BY International License.] As always, many questions remain to be answered. In particular, it is not clear why acute attacks in human patients involve both sensory and motor nerves, while chronic neuropathy predominantly affects motor nerves in humans as well as mice. [Preferential involvement of motor axons in the mouse model can be attributed to differences in macrophage activation between sensory and motor nerves; however, given that increased SARM1 activity is observed in both nerve types, it is not clear what leads to these nerve-specific differences in macrophage activation. It is also possible that motor neurons are particularly vulnerable to SARM1 activation; in agreement with that possibility, hypermorphic SARM1 variants were found to be enriched in patients with amyotrophic lateral sclerosis (ALS); (Gilley et al., 2021; Bloom et al., 2022)]. Moreover, it is not understood why NMNAT2 deficiency primarily affects peripheral nerves in both mice and humans when SARM1 acts as the central regulator of Wallerian degeneration in both CNS and PNS; a plausible explanation that needs experimental confirmation is that longer length of PNS axons makes them more vulnerable to NMNAT2 deficiency. Finally, the prodegenerative effect of macrophage activation in the Nmnat2V98M/R232Q mouse model was entirely unexpected, not only because it was previously thought that SARM1 acts in an entirely cell-autonomous manner, but also because of a large body of prior research that demonstrated a key role of macrophages in axon regeneration [as discussed in advance #1 of my last update (Margeta, 2022)]. These discrepancies suggest that macrophage effects on axonal degeneration are complex and context-dependent, and raise the possibility that macrophage depletion therapies could be either beneficial or detrimental, depending on the specific clinical scenario. One intriguing possibility, which needs to be addressed by future studies, is that macrophage activation promotes degeneration of damaged but still viable axons in chronic axonal neuropathies, while promoting axonal regeneration following severe insults (such as nerve trauma or ischemia) that result in catastrophic axonal injury. Advances in understanding of etiology and pathogenesis of neuromuscular diseases 5. Axonal injury in Guillain-Barré syndrome starts at the nodes of Ranvier GBS is an autoimmune disorder of the peripheral nerves and spinal roots that is typically triggered by a preceding infection or vaccination; nerve injury is the result of autoantibody-induced complement fixation and macrophage activation. While GBS is traditionally considered to be a demyelinating disease (also known as acute inflammatory demyelinating polyneuropathy / AIDP), it can also manifest as a primary acute motor or sensorimotor axonal neuropathy (AMAN / AMSAN). Bystander axonal injury can also develop in the AIDP variant of GBS, secondary to Schwann cell (SC) injury. Regardless of the disease variant, the long-term severity of GBS is ultimately determined by the extent of axon loss, which is less reversible than the myelin loss; however, the mechanisms that underlie the primary and secondary axon injury in GBS have been difficult to dissect because the key autoantibody targets (such as GM1 gangliosides) are often expressed by both neurons and SCs. To circumvent this challenge, a research group in Scotland created model mice that selectively express GM1 ganglioside on either SC or neuronal membranes; the peripheral nerves of these mice were then exposed to monoclonal anti-GM1 antibodies and normal human serum (as a source of complement) to trigger acute or subacute nerve injury, either ex vivo (in the triangularis sterni nerve-muscle preparations) or in vivo (McGonigal et al., 2022). In both mouse models, early injury [following a short (4 h) treatment with anti-GM1 antibodies and complement] occurred at the nodes of Ranvier (NoR); this is concordant with the neuropathologic studies of patients with GBS, which demonstrated nodal autoantibody and complement deposition (Hafer-Macko et al., 1996a; Hafer-Macko et al., 1996b) along with alterations of the nodal ultrastructure (Griffin et al., 1996; Vallat et al., 2020). In Neuronal GBS model mice (which express GM1 selectively in neurons), formation of the membrane attack complex (MAC) pores in the nodal axolemma led to calpain-mediated cleavage of adaptor protein ankyrin-G, dispersion and disappearance of the nodal sodium channel clusters, and conduction failure; however, there were no structural changes in the paranodal SC loops and no disruption of the cell adhesion complexes that form “transverse bands” at the axo-glial interface. In Glial GBS model mice (which express GM1 selectively in SCs), injury started with the MAC-induced distortion of the paranodal SC loops and calpain-mediated cleavage of the SC scaffolding protein ankyrin-B, leading to disruption of transverse bands, dispersion of the sodium channel clusters, and conduction failure (Fig. 4). Thus, the ultimate functional consequence – acute conduction failure – was the same in both mouse models, although the underlying disease mechanisms and associated structural changes were different. Acute conduction failure – which is seen in many human GBS patients in addition to these experimental disease models – is often temporary and reversible; however, it can also lead to subsequent axonal transection / Wallerian degeneration. Indeed, extended (20 h) treatment with anti-GM1 antibodies and complement in vivo led to a loss of the distal motor axon integrity in Glial mice, indicative of secondary / bystander axonal degeneration due to nodal SC injury. (The authors have yet to perform similar extended treatment studies in their Neuronal mice.) Taken together, these results point to NoR as the site of initial injury in both neuronal/axonal and glial/demyelinating forms of GBS. [Interestingly, NoR is also the main site of injury in autoimmune nodopathies, which are caused by antibodies that directly target glial or neuronal cell-adhesion molecules expressed in the nodal membranes; clinically, autoimmune nodopathies show significant overlap with GBS, but are typically more severe and resistant to IVIg immunomodulatory therapy (Martin-Aguilar et al., 2022).]
Figure 4. Ultrastructural evaluation of diaphragms from in vivo GBS models. Neuronal and Glial mice were dosed i.p. with 50 mg/kg anti-GM1 Ab followed 16 hours later with 30 μL/g normal human serum (NHS) (injury) or NHS only (control). (A) A normal paranode from Glial control tissue. This image is also representative of the Neuronal control NoR (not shown). (B) Higher magnification of boxed region from A shows tight junctions (large arrowhead) between the paranodal loops, and transverse bands (TBs, small arrowheads) at the axo-glial junction between the axon and paranodal loops. (C) Injured Glial NoRs show severely disrupted paranodal loop organization compared with control. (D) Magnification of boxed area from C, shows TBs are present between the paranodal loops and axon at the juxtaparanodal-proximal paranode (above black line); however, they are absent at the node-proximal border (above white line, right of asterisk). (E) Injured Neuronal NoRs show no architectural disruption. (F) Neuronal control motor nerve terminal (MNT) displays normal architecture and contains synaptic vesicles (black arrows). (G) Disturbance to the injured Neuronal MNT includes an absence of neurofilament, synaptic vesicles, and the formation of dense or vacuolated mitochondria (white arrows). Results are representative of analysis from 8–10 NoRs per mouse (n = 3/genotype/treatment). [This figure and its legend were adopted from Figure 6 in (McGonigal et al., 2022); this use is permitted under the Creative Commons Attribution 4.0 CC-BY International License.] What is the relevance of these findings for human GBS? Since GM1 gangliosides (and other ganglioside antigens) are expressed by neurons as well as SCs, both of these mechanisms are likely in play in human disease, and which mechanism dominates in any given GBS patient likely depends on a combination of factors, including antibody titer / specificity and intrinsic host factors that shape disease vulnerability. Moreover, these findings have potential diagnostic implications: taken together with the 2020 ultrastructural study of peripheral nerves from patients with GBS and other nodo-paranodopathies (Vallat et al., 2020), which showed ultrastructural nodal pathology similar to ultrastructural abnormalities seen in Glial GBS mice (Fig. 4), they suggest that electron microscopic evaluation of NoR could help delineate immune-mediated from ischemic and other causes of axonal injury, and should therefore be included in the nerve biopsy workup whenever an obvious cause of axon degeneration (such as vasculitis) is not detected in a nerve biopsy. 6. Target formation is indicative of muscle fiber reinnervation Target fibers are one of the classic diagnostic features of a neurogenic change secondary to lower motor neuron (LMN) injury. Targets are most commonly seen in type 1 fibers, are best visualized on oxidative stains, and in the cross-sectional view consist of several distinct concentric layers / zones – typically a central pale area devoid of mitochondrial activity and reminiscent of a core, an intermediate zone that shows a decreased but not entirely absent mitochondrial activity, and an outer zone that resembles the rest of the myofiber and often shows an increase in mitochondrial activity (Fig. 5A-B). Ultrastructurally, the central zone contains the Z-disc material and is devoid of glycogen and mitochondria, the intermediate zone shows disorganized myofilaments with mild Z-line abnormalities, while the outer zone contains ultrastructurally normal myofilaments. Similar formations that have a less distinctively layered appearance are called “targetoid” and can be more difficult to distinguish from cores if they are not accompanied by true targets and/or other diagnostic features of neurogenic change. However, targets are not present in all muscle biopsies with neuropathic changes, and can be seen both in small angulated and normally sized fibers. As a result, it has been unclear whether target formation reflects denervation [as originally proposed (Engel, 1961)] or reinnervation [as suggested by subsequent animal model studies (De Reuck et al., 1977)]. A recent proteomic study done by the Kley lab sheds a new light on this old question, providing compelling evidence that target formation reflects myofibril assembly that occurs following reinnervation (Krause et al., 2022).
Figure 5. Target fibers. (A) NADH-TR stain shows two type 1 fibers with central target formations; on myosin heavy chain immunohistochemistry, these fibers were positive for MyHC-slow (MHC7; not shown). The same microscopic field, captured on consecutive cryosections, is shown in all main figure panels; target formations are highlighted by arrowheads. (B) The multilayered appearance of target formations (arrowheads), with the central pale zone, dark mitochondria-rich outer rim, and an intermediate zone in between, is more obvious on SDH histochemistry. (C) Desmin immunohistochemistry highlights the inner and intermediate zones of target formations, where new myofibrils are assembled; in some cases, desmin staining is most prominent in the intermediate zone (main panel), while in others it is equally prominent in both zones (inset). (Orange line, inner zone boundary; green line, intermediate zone boundary; black line, outer zone boundary.) (D). LC3 immunohistochemistry highlights autophagosomes, a key component of the quality-control machinery responsible for degradation of misassembled or damaged myofibrils; in some target formations, LC3 staining is most prominent in the central zone (main panel), while in others it is most prominent in the intermediate zone (inset). Different patterns of target formation staining observed with desmin and LC3 immunostains could reflect different stages in myofibril assembly following fiber reinnervation. Scale bar, 50 µm. To investigate the molecular composition of targets, Krause and co-authors employed a non-biased, hypothesis-free proteomic strategy: they used 20 muscle biopsies from patients with neurogenic muscle atrophy to micro-dissect targets and analogously sized portions of uninvolved type 1 fibers, and then compared the protein composition of both samples via label-free mass spectroscopy. Out of 1026 proteins identified in these samples, 55 proteins were overrepresented and 40 proteins underrepresented in targets compared to the control regions of type 1 fibers. Unsurprisingly, the underrepresented proteins were mainly mitochondrial proteins. The overrepresented proteins were more interesting and could be classified into four main groups based on their subcellular localization and/or function: (i) Z-disc and actin dynamics (15 proteins, including filamin C and desmin), (ii) myosins and myosin-associated proteins (10), (iii) protein biosynthesis (10, including ribosomal proteins), and (iv) molecular chaperones (5, including alpha(B)-crystallin and BAG3); the remaining 15 proteins were classified as “other.” The key mass spectroscopy findings were then validated by immunofluorescent staining and showed zonal pattern of expression, with filamin C, other Z-disc proteins, and chaperones in the central zone, desmin mainly in the intermediate zone (Fig. 5C), and myosins in both zones. The model that emerged from these proteomic findings suggests that the central and intermediate zones are the sites of new myofibril synthesis; premyofibrils are formed in the central zone, and then assembled and linked with the existing myofibrils in the intermediate zone. Chaperones and other components of the machinery involved in chaperone-assisted selective autophagy (such as autophagosomal protein LC3; Fig. 5D) are enriched in both zones, providing a quality-control mechanism that ensures that nascent myofibrils are either properly built and assembled or immediately degraded. While this study represents a major advance in our understanding of the reinnervation-induced myofiber rebuilding mechanisms, some aspects of this process still remain to be elucidated. For example, it is not clear why targets are mainly seen in type 1 fibers. Does the same process occur in a less structured / morphologically less distinctive way in type 2 fibers, or are reinnervated type 2 fibers rebuilt through entirely different molecular mechanisms? Furthermore, it remains to be established how the motor neuron axon and muscle fiber interact to orchestrate this highly organized process of new myofibril formation. 7. Sporadic inclusion body myositis revisited: What comes first, inflammation or myodegeneration? Sporadic inclusion body myositis (sIBM) primarily affects people older than 50 and has distinctive clinicopathologic features that differentiate it from other idiopathic inflammatory myopathies; clinically, there is predominant involvement of quadriceps and finger flexor muscles, while pathologically, there is endomysial inflammation accompanied by diffuse MHC-I upregulation and invasion of intact muscle fibers by cytotoxic CD8+ T lymphocytes and macrophages, along with mitochondrial abnormalities, chronic myopathic features resembling muscular dystrophy, rimmed vacuoles, and p62- and TDP-43-positive protein aggregates. However, pathogenesis of sIBM remains poorly understood. In particular, the role of inflammation has remained controversial: although sIBM muscle biopsies typically show prominent inflammatory infiltrates, the disease does not respond to immunosuppression (or any other therapy) and generally leads to wheelchair dependence ~15 years following the diagnosis. Several important studies published in 2019 [and discussed in the first paper in this review series (Margeta, 2020)] suggested that the lack of response to immunomodulatory therapy is due to involvement of terminally differentiated effector memory T cells (so-called TEMRA cells), which are positive for KLRG1 (killer cell lectin-like receptor G1; a marker of T cell exhaustion) and resistant to steroid-induced apoptosis (Greenberg et al., 2019; Knauss et al., 2019). Several important follow-up studies were published in 2022, prompting me to revisit this important topic in the current review. Our understanding of sIBM pathogenesis has been limited by the lack of a disease model that replicates both inflammatory and myodegenerative features of this disease. To circumvent this barrier, Britson and colleagues developed a xenograft model of sIBM, in which human muscle biopsy tissue is transplanted into immunodeficient mice; following transplantation, transacted mature muscle fibers degenerate and are replaced by newly formed fibers generated from satellite cells that were harnessed along with the graft (Britson et al., 2022). Remarkably, in sIBM xenografts – but not in the xenografts generated from relatively normal muscle biopsies or from biopsies showing other types of myositis – regenerated myofibers showed typical features of sIBM, including MHC-I positivity, rimmed vacuoles, loss of nuclear TDP-43 expression, and mitochondrial abnormalities (COX deficiency). In addition, sIBM xenografts contained donor-derived oligoclonal CD8+KLRG1+ TEMRA cells that were enriched compared to the source sIBM biopsies. Finally, ~50% of sIBM xenografts (but only 3% of control xenografts) showed expression of cryptic exons in TDP-43-regulated mRNAs, indicative of functional TDP-43 deficiency. (TDP-43 is an RNA-binding protein that maintains the health of the transcriptome by repressing incorporation of non-conserved cryptic exons during pre-mRNA splicing; cryptic exon expression in the TDP-43-regulated, muscle-specific mRNA transcripts was 84% sensitive and 99% specific marker of sIBM in the cohort of 119 myositis patients enrolled in this study). Having developed the xenograft sIBM model, Britson et al. used to it to evaluate whether treatment with monoclonal anti-CD3 antibody OKT3 would improve various aspects of sIBM pathology. The OKT3 treatment, which almost completely depleted T cells and significantly reduced the number of KLRG1-positive cells in sIBM xenografts after 4 months of weekly infusions, led to a significant decrease in the extent of MHC-I upregulation; however, it had no significant effect on myodegenerative sIBM features (COX-negative fibers, rimmed vacuoles, and p62-positive protein aggregates). In addition, there was no change in the aberrant expression of cryptic exons. These results quite definitively demonstrate that myodegenerative pathology is intrinsic to sIBM muscle and can persist independent of sustained inflammation or circulating factors, providing an additional explanation for refractoriness of sIBM to standard immunosuppressive treatments, but also diminishing hope that therapies that directly target KLRG1-positive TEMRA cells (which are already in development) will significantly benefit sIBM patients. However, it is less clear what these findings mean for our understanding of sIBM pathogenesis: while the authors of this study favor a model in which TDP-43 dysfunction is an early event in the disease progression that then drives aberrant mRNA splicing, neoantigen expression, and secondary inflammation, it is equally possible that chronic inflammation within the human xenograft donor prior to the biopsy (and/or within the early xenograft, prior to the OKT treatment) leads to genetic and/or epigenetic alterations in satellite cells, which then recapitulate TDP-43 dysfunction and myodegenerative changes in newly formed muscle fibers. Additional work will be required to distinguish between these two models of sIBM pathogenesis; in particular, it should be established whether aberrant expression of TDP-43-regulated cryptic exons is also present in hereditary inclusion body myopathies, which show sIBM-like myodegenerative features but lack inflammation. Interestingly, the results of another 2022 paper provide support for inflammation as an early event in sIBM disease progression (Kleefeld et al., 2022). In their cross-sectional study, Kleefeld and colleagues examined the similarities and differences between polymyositis with mitochondrial pathology (PM-Mito) and sIBM. In contrast to sIBM, which has a distinctive clinical phenotype, PM-Mito is not a clinically well-defined entity; pathologically, however, it shows some sIBM-like features (lymphocytic endomysial inflammation, muscle fiber invasion by inflammatory cells, and mitochondrial pathology / COX-deficient fibers) but lacks rimmed vacuoles, chronic myopathic changes, and p62- or TDP-43-positive protein aggregates (Blume et al., 1997; Hiniker et al., 2013). Although most patients with PM-Mito over time progress to sIBM, at least partial response to immunosuppression has been observed in a subset of cases (Levine and Pestronk, 1998; Winkler et al., 2021), raising the possibility that PM-Mito and sIBM exist on a spectrum, with PM-Mito an early – and potentially treatable – stage of sIBM. The findings reported by Kleefeld et al. support this model: they found an essentially identical inflammatory molecular signature in PM-Mito and sIBM biopsies, with greater activation of type II than type I interferon response and expression of cryptic exons in both sets of cases (although frequency of cryptic exon expression was greater in sIBM than PM-Mito biopsies). In contrast to these similarities, KLRG1 and GBP6 (guanylate binding protein family member 6) genes were differentially expressed, with both KLRG1 and GBP6 mRNA upregulation detectable only in sIBM biopsies. In agreement with this finding, GBP6-positive macrophages, GBP6-expressing myofibers, and KLRG1-positive T cells were significantly more abundant in sIBM than PM-Mito samples. Overall, most differences between PM-Mito and sIBM biopsies were quantitative rather than qualitative, with a gradient of severity observed even within the PM-Mito group (which could be histologically divided into “mild”, “typical”, and “pre-IBM” subgroups). Importantly, this progression of severity was also observed in serial biopsies from two individuals, the first showing PM-Mito and the second (5-7 years later) sIBM. Based on these findings, Kleefeld and colleagues recommended that PM-Mito should be renamed early sIBM and that both disorders together should be classified as “sIBM-spectrum disease” (sIBM-SD). In addition, they suggested that the difference in KLRG1 expression between early and late sIBM stages reflected disease duration, with emergence of exhausted T cells a consequence of prolonged inflammation / antigenic stimulation and a harbinger of transition into a therapy-resistant end stage of sIBM-SD. In agreement with that hypothesis, another recent study used “deep immunophenotyping” of blood T and NK cells from sIBM patients to show that increased CD8+ T cell differentiation correlated with sIBM duration (Goyal et al., 2022). Among many questions that still need to be answered is the “status” of polymyositis without mitochondrial abnormalities, which is histologically identical to PM-Mito except for the absence of ragged red and COX-negative fibers (Hiniker et al., 2013). It is not currently known whether polymyositis is a separate disease, or an even earlier stage of sIBM-SD that can be effectively treated by standard immunosuppressive therapy and is therefore less likely to progress to a chronic inflammation-driven, treatment-resistant end stage. Given that inflammation can lead to secondary mitochondrial abnormalities in other inflammatory myopathies (Hedberg-Oldfors et al., 2022), it is possible that emergence of COX-negative fibers in PM-Mito represents the first step on the pathway towards treatment resistance in the setting of persistent chronic inflammation. Advances in neuromuscular disease diagnostics 8. Improved genetic testing techniques for facioscapulohumeral and myotonic muscular dystrophies Despite widespread availability and relatively low cost of advanced genetic testing techniques (such as syndrome-specific multigene panels and whole exome sequencing), 50-75% of presumed genetic neuromuscular disease cases currently remain undiagnosed (Cummings et al., 2017). The underlying reasons for these diagnostic challenges are complex and varied, but include the inability of these sequencing techniques to detect structural rearrangements and copy number variants. Fortunately, two new genetic testing strategies developed in 2022 will alleviate some of these challenges, simplifying the diagnosis of two common muscular dystrophies (Erdmann et al., 2022; Stevanovski et al., 2022). Facioscapulohumeral muscular dystrophy (FSHD) is the third most common muscular dystrophy; it has a fairly unique clinical presentation, with the facial and shoulder girdle weakness that progresses to involve distal legs and the pelvic girdle. However, the age of onset is highly variable (from infancy to late adulthood) and the phenotype can also vary, making the diagnosis challenging. Histopathologic features of FSHD are relatively unique (Fig. 6), but not sufficiently specific to enable definitive diagnosis; hence, directed genetic testing is generally required at some point of diagnostic work-up. Molecularly, FSHD is caused by aberrant expression of DUX4 (double homeobox 4) protein in skeletal muscle, which has myotoxic effects. DUX4 is a transcription factor that regulates expression of genes important for pre- and post-implantation development, but is epigenetically silenced in most adult tissues except in thymus and testis, where it has an unknown function (Mocciaro et al., 2021). In FSHD, there is a loss of DUX4 repression, but that derepression occurs through mechanisms that differ between two disease subtypes, FSHD1 and FSHD2. In FSHD1 (~95% of cases), DUX4 derepression is caused by contraction of the D4Z4 macrosatellite repeat array in the subtelomeric region of chromosome 4q35 to <12 repeating units; in FSHD2 (~5% of cases), there is global hypomethylation of the D4Z4 array due to mutations in one of the several genes that encode proteins required for epigenetic suppression. (A permissive haplotype 4qA or 4qAL, which provides a polyA signal for DUX4 mRNA, is required for phenotypic disease expression in either disease subtype.) Currently, genetic diagnosis of FSHD requires (i) confirmation of the permissive haplotype, (ii) determination of the D4Z4 repeat length, typically via Southern blotting (which requires a large amount of high molecular weight DNA), and (iii) sequencing of epigenetic suppressor genes if the D4Z4 repeat length is found to be within normal limits. The new, much more streamlined approach for FSHD testing developed by Erdmann et al. is based on methylation profiling: following confirmation of the permissive haplotype, a high-throughput methylation profile analysis is used to determine the global methylation level of the entire D4Z4 repeat array as well as the regional methylation of its most distal repeat unit; individuals with isolated distal hypomethylation have FSHD1, while individuals with both global and distal hypomethylation have FSHD2 (Erdmann et al., 2022). Not only does this new diagnostic approach require much smaller quantity of DNA and less laboratory effort, it also detects cases that are missed with traditional methods (for example, patients with complex chromosomal rearrangements that cannot be detected by Southern blotting) or cases that are in the gray zone between FSHD1 and FSHD2 (which represent the two ends of the epigenetic disease continuum). In addition, Erdmann et al. have shown that the D4Z4 repeat methylation profile is a biomarker of FSHD severity: in their cohort of 148 patients, the distal methylation level was better correlated with clinical findings (including the age of onset) than the D4Z4 repeat length. Finally, this study provides definitive evidence that FSHD pathogenesis is ultimately driven by epigenetic rather than genetic mechanisms.
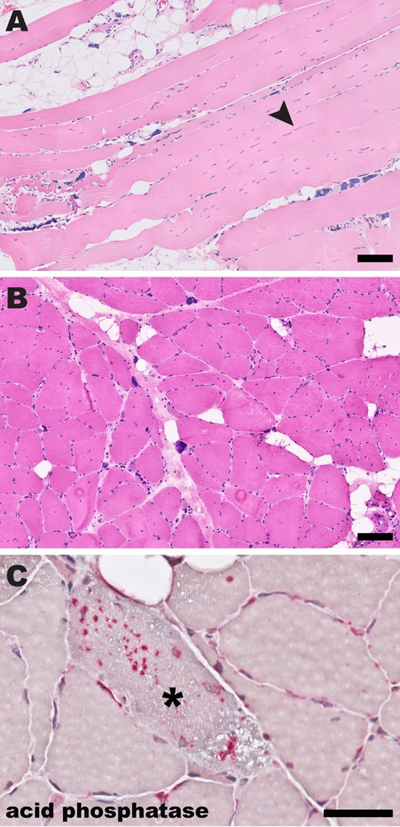
Figure 6. Histologic findings in facioscapulohumeral dystrophy (FSHD). (A) A representative H&E-stained section shows fiber size variation, endomysial fibrosis, and perimysial inflammation that focally extends into the endomysium. (B) Moderately frequent lobulated fibers (arrowheads) are best seen on NADH-TR histochemistry. (C) MHC-I is diffusely upregulated in muscle fibers. (D) CD31 immunohistochemistry highlights enlarged endomysial capillaries. Based on the biopsy findings, the patient underwent genetic testing for FSHD that showed pathogenic D4Z4 repeat contraction (8 repeats) detected on a 4QA permissive haplotype, diagnostic of FSHD1. All stains were performed on cryosections; scale bar, 50 µm. Myotonic dystrophy (dystrophia myotonica; DM) is the most common muscular dystrophy in adults and the second most common muscular dystrophy overall. As with FSHD, there are two distinct DM subtypes, DM1 (which accounts for >95% of cases) and DM2 (which is much rarer). Although their clinical features and causative genes differ, both DM1 and DM2 are short tandem repeat (STR) expansion diseases: DM1 is caused by expansion of the CTG repeat in the 3’ untranslated region of DMPK gene, while DM2 is caused by expansion of the CCTG repeat in intron 1 of CNBP/ZNF9 gene. Interestingly, the mechanism of myotonia is the same in both DM subtypes: mutant RNAs sequester RNA-binding and processing proteins, leading to incorrect splicing of ClC-1 chloride channel (which is required for the maintenance of normal resting membrane potential). Histopathology of DM1 and DM2 is strikingly similar, with very high internal nucleation and frequent subsarcolemmal nuclear aggregates in the setting of usual dystrophic changes (Fig. 7); therefore, the definitive diagnosis requires genetic testing. Although small STR expansions (< 300 bp) can be detected by conventional PCR, genetic diagnosis of most STR expansion disorders (which in addition to DM1 and DM2 include Huntington’s disease, fragile X syndrome, hereditary cerebellar ataxias, C9orf72-related ALS / frontotemporal degeneration, and other neurologic diseases) currently relies on Southern blotting and/or tandem repeat-primed PCR, and therefore faces many challenges already described for FSHD in addition to requiring separate probes and/or primers for each STR. These challenges are either solved or bypassed by the new genetic strategy developed by Stevanovski et al., which relies on programmable targeted long-read nanopore sequencing to genotype all known neuropathogenic STRs in parallel, using a single molecular assay (Stevanovski et al., 2022). One particularly appealing feature of the platform used for this assay is that it permits flexible inclusion of targets, so that a specific assay used for any given patient can be individually tailored based on the clinician’s input and/or the patient’s preferences; another is that sequencing can be combined with methylation profiling, providing epigenetic as well as genetic information in a single step. As a result, this new genetic testing approach will likely advance basic research in addition to patient care: much remains to be learned about the fundamental biology of the STR expansion disorders and the key variables that distinguish normal and disease-causing alleles, and the availability of a relatively inexpensive high-resolution molecular STR assay should accelerate research progress in all these areas.
Figure 7. Histologic findings in myotonic dystrophy (DM). (A) A representative H&E-stained formalin-fixed, paraffin embedded section shows fiber size variation, endomysial fibrosis with fatty infiltration, frequent subsarcolemmal nuclear aggregates, and nuclei running in chains along the longitudinal fiber axis (arrowhead). (B) An H&E-stained cryosection shows similar findings, with prominent subsarcolemmal nuclear aggregates and a marked increase in internal nuclei. (C) An acid phosphatase-stained cryosection shows a fiber with prominent sarcoplasmic acid phosphatase-positive granules (asterisk). Based on the biopsy findings, the patient underwent genetic testing for myotonic dystrophy that showed >15600 repeats in one allele of CNBP/ZNF9 gene, diagnostic of DM2. (Histologic findings in DM1 are very similar.) Scale bars: A and B, 100 µm; C, 50 µm. 9. Accumulation of phosphorylated TDP-43 in motor nerves and intramuscular nerve twigs is a diagnostic marker of amyotrophic lateral sclerosis ALS is a neurodegenerative disease that typically affects both upper and lower motor neurons and is the most common cause of motor neuron disease (MND) in adults; pathologically, it is characterized by selective motor neuron loss that is accompanied by aggregates of phosphorylated TDP-43 (pTDP-43) in the remaining motor neurons and glia. During life, ALS diagnosis is based on the clinical criteria; however, there is a significant phenotypic heterogeneity among ALS patients, and up to 20% of patients with an LMN-predominant form of ALS are misdiagnosed prior to death. Based on the recent work from two separate research groups in Italy and Japan, immunohistochemical detection of pTDP-43 aggregates in motor nerve biopsies (Riva et al., 2022) and/or motor nerve twigs in muscle biopsies (Kurashige et al., 2022) may be a useful tool for closure of that diagnostic gap. Riva and colleagues performed a retrospective cohort study to assess whether TDP-43 accumulation in motor nerve biopsies can serve as a useful biomarker of ALS. They retrospectively reviewed the clinical course of 113 patients who underwent a diagnostic biopsy of the anterior motor branch of the obturator nerve, typically for evaluation of a diagnostically confusing LMN syndrome, during the 25-year period between 1994 and 2019. Of these 113 patients, 102 had sufficient clinical data to be included in the study; 71 (69.6%) were ultimately diagnosed with ALS. In the first part of this study, the authors performed blinded histopathology review of original slides for all 102 study participants; 61 biopsies were diagnosed as pathologic MND, 16 were diagnosed as pathologic motor neuropathy, while 25 were non-diagnostic. Excluding the cases with pathologic diagnosis of motor neuropathy (which had 100% sensitivity and 100% specificity for the ultimate diagnosis of motor neuropathy), the specificity of pathologic MND pattern for the final diagnosis of ALS in that patient cohort was 66.6%, while its sensitivity was 78.9%. (Interestingly, the degree of axon loss in ALS cases was predictive of the overall and post-biopsy survival length.) In the second part of the study, immunohistochemistry with polyclonal anti-TDP-43 and anti-p(S409/410)TDP-43 antibodies was performed on biopsies from 80 patients for which sufficient archival tissue was available; in this subcohort, 57 patients (71.3%) were ultimately diagnosed with ALS. Accumulation of TDP-43 in myelinated axons and SC cytoplasm was more commonly detected in ALS patients than in non-ALS patients, but this difference was statistically significant only when anti-pTDP-43 antibodies were used; overall, the presence of pTDP-43 aggregates had the specificity of 65.2% and sensitivity of 98.2% for the final diagnosis of ALS. Interestingly, some of the patients with a false-positive pTDP-43 signal in their motor nerve biopsies were ultimately diagnosed with inclusion body myositis or idiopathic motor neuropathy. While this finding represents a limitation from the diagnostic perspective, it highlights the mechanistic connections between these disorders: multisystem proteinopathies, which clinically manifest as one or more of these diseases, have been etiologically linked to impaired biology of TDP-43-containing RNA stress granules (Korb et al., 2021). Interestingly, axonal pTDP-43 aggregates were detected in 11 of 11 ALS cases with normal (and therefore nondiagnostic) motor nerve biopsies, suggesting that pTDP-43 accumulation is an early event in the disease progression that precedes LMN degeneration and axon loss. One limitation of the current study is that the authors did not evaluate whether specificity and sensitivity of the motor nerve biopsy in the ALS diagnosis could be improved by combining the results of standard histopathologic analysis with pTDP-43 immunohistochemistry; this important question should be assessed in future studies from this or other research groups. Kurashige et al. approached the same question in a slightly different way, by focusing on pTDP-43 accumulation in the intramuscular motor nerve twigs (Kurashige et al., 2022). In the first part of their investigation, these authors performed an autopsy case-control study that included 10 patients with autopsy-confirmed sporadic ALS and 12 control, non-ALS patients; for each case, muscle samples were obtained from the tongue, diaphragm, biceps brachii, and rectus femoris. The second part of the project was a retrospective cohort study, which started by screening 450 patients who underwent a diagnostic muscle biopsy at the authors’ institutions between 2004 and 2019; after all patients with a myopathy and patients with pathogenic variants in the ALS-associated genes were excluded, the study cohort consisted of 114 patients, 71 of whom had biopsies that included intramuscular nerve twigs. All study samples were stained with both polyclonal and monoclonal anti-p(S409/410)TDP-43 antibodies, which gave identical results. In autopsy cases, axonal pTDP-43-positive aggregates (but no FUS-, p62-, or ubiquitin-positive aggregates) were detected in all ALS patients but not in any of the controls (Fig. 8); ~50% of nerve twigs were involved in any given case, with no significant differences among the four sampled muscle groups. In the biopsy portion of the study, 33 of 71 cases with intramuscular nerve twigs (46.5%) showed axonal pTDP-43-positive aggregates; all these patients were ultimately diagnosed with ALS (9 with the LMN-predominant form), while none of 38 pTDP-43-negative patients received the ALS diagnosis, and were instead ultimately diagnosed with other MNDs or neuropathies. (Interestingly, only 4 of the 114 patients in the muscle biopsy cohort were clinically suspected of having ALS prior to the biopsy.)
Figure 8. Axonal pTDP-43–positive accumulations in intramuscular nerve bundles of skeletal muscle in the postmortem case-control study. (A and D) H&E-staining in axons of intramuscular nerve bundles showed no differences between patients with amyotrophic lateral sclerosis (ALS) and those without ALS except for the density of intramuscular nerves (original magnification: A, ×40; D, ×60). (B) Immunohistochemical analysis with mouse monoclonal pTDP-43–positive accumulations in intramuscular nerve bundles (arrowheads) of patients with spontaneous ALS (SALS) (original magnification, ×40). (E) Immunostaining with mouse monoclonal antibody against pTDP-43 in patients with non-ALS diseases did not reveal any abnormalities in intramuscular nerve bundles (original magnification, ×40). (C) Immunohistochemical analysis with rabbit polyclonal pTDP-43 antibody also revealed pTDP-43–positive accumulations in intramuscular nerve bundles (arrowheads) of patients with SALS (original magnification, ×60). (F) Immunostaining with rabbit polyclonal antibody against pTDP-43 in patients with non-ALS diseases did not reveal any abnormalities in intramuscular nerve bundles (original magnification, ×40). [This figure and its legend were adopted from Figure 2 in (Kurashige et al., 2022); this use is permitted under the Creative Commons Attribution 4.0 CC-BY International License.] Taken together, these two studies provide compelling evidence that immunohistochemical detection of pTDP-43-positive protein aggregates in motor axons is a useful diagnostic marker of ALS. While the results of both studies need to be replicated by other researchers, these findings suggest that muscle biopsy evaluation is a particularly promising diagnostic approach: not only are muscle biopsies easier to do and have lower morbidity than motor nerve biopsies, they also showed higher overall specificity and sensitivity than nerve biopsies (perhaps because potentially confounding myopathic biopsies could easily be excluded). Of course, the utility of muscle biopsies for ALS diagnosis is limited by the lack of intramuscular nerve twigs in many specimens (~40% in this study cohort); obtaining larger tissue samples and performing serial level sections will probably help decrease the number of inadequate biopsies if this evaluation becomes a standard component of the ALS diagnostic work-up. Advances in neuromuscular disease treatment 10. SARM1 inhibitors block Wallerian degeneration in preclinical models of axonal neuropathy As discussed in advance #4, SARM1 is the central regulator of intrinsic axon destruction program / Wallerian degeneration; deletion of SARM1 in mice prevents axonal degeneration in both CNS (for example, in mouse models of traumatic brain injury) and PNS (in mouse models of axotomy and chemotherapy-induced peripheral neuropathy). The central role of SARM1 in Wallerian degeneration raised the possibility that SARM1 blockade would benefit patients with a broad spectrum of neurologic disorders, leading to a major effort by multiple different research groups (both in academia and biotechnology companies) to develop small molecule inhibitors of SARM1. Initial proof-of-principle studies with catalytic site SARM1 inhibitors were published in 2021 (Bosanac et al., 2021; Hughes et al., 2021), with more comprehensive preclinical studies of catalytic inhibitors (Bratkowski et al., 2022; Shi et al., 2022) and a discovery of a new class of allosteric SARM1 inhibitors (Feldman et al., 2022) reported in 2022. SARM1 is a multi-domain enzyme that functions as an oligomer; it contains an auto-inhibitory N-terminal armadillo repeat (ARM) domain, repeat sterile alpha motif (SAM) domains that are required for dimerization / oligomerization, and a catalytic C-terminal domain that includes the toll/interleukin receptor (TIR) motif. The TIR domain contains the NAD+ hydrolase activity that catabolizes NAD+ into nicotinamide, adenosine diphosphate ribose (ADPR), and cyclic ADPR (which is the second messenger that triggers the Wallerian degeneration pathway). Under normal conditions, NAD+ is bound to the ARM domain, resulting in autoinhibition of the SARM1 activity; when the NMN/NAD+ ratio increases (typically due to a decreased production of NAD+ by NMNAT2), NMN binds to the ARM domain instead of NAD+, resulting in a conformational change that leads to oligomerization of the TIR domain and relief of SARM 1 autoinhibition (Feldman et al., 2022). Several groups have developed non-competitive, adduct-forming small molecule inhibitors of the SARM1 NAD+ hydrolase site, with most comprehensive characterization to date reported by Bratkowski and colleagues. These non-competitive SARM1 inhibitors function as pseudo-substrates / pro-drugs that intercept NAD+ hydrolysis and form inhibitor-ADPR adducts, which act as highly potent inhibitors of the catalytic site, with slow on and off rates and the dissociation constant in the low nanomolar range. While all studies have shown that these catalytic site inhibitors prevent axonal degeneration in vitro, Bratkowski et al. went beyond cellular axon injury models: they used preclinical mouse models of acute peripheral nerve injury (a sciatic nerve transection model and a model of vincristine-induced peripheral neuropathy) to demonstrate that these drugs are both safe and effective in vivo, with efficacy approaching the efficacy of genetic SARM1 deletion. Specifically, treatment with NB-3 (the compound with chemical properties most suitable for in vivo use) suppressed an increase in the plasma level of neurofilament light chain (a biomarker of axonal injury) and led to preservation of distal nerve fiber integrity in both acute nerve injury models; in addition, it attenuated toxin-induced allodynia / neuropathic pain in the vincristine model. (The transection model cannot be used for functional studies because connections between the CNS and periphery are completely disrupted.) Critically, the NB-3 treatment was effective not only when it was administered prophylactically but also when it was given several hours after injury, a treatment paradigm that is more relevant for potential clinical use. In a separate line of the SARM1 inhibitor research, Feldman and colleagues used a chemical proteomic approach to discover an entirely novel class of SARM1 inhibitors: these electrophilic compounds (tryptoline acrylamides) target cysteine C311 in the autoregulatory ARM domain, resulting in allosteric inhibition of SARM1 enzymatic activity (Feldman et al., 2022). Thus far, these new compounds were evaluated only in the cellular models of axon degeneration, where their efficacy was similar to the efficacy of catalytic site inhibitors; it remains to be determined whether they will be similarly safe and effective in vivo. Regardless of the in vivo effectiveness of these specific compounds, this study provides initial evidence that SARM1 can be pharmacologically inhibited through allosteric as well as enzymatic mechanisms; because different classes of SARM1 inhibitors are likely to have different side effects and different optimal clinical uses, this finding is both important and exciting. Where is this research headed? First, there should be additional preclinical testing of all compounds discovered to date using models of chronic axonal injury (including the NMNAT2 deficiency model described in advance #4) that more closely resemble clinical scenarios where SARM1 inhibitors will be used. [In the setting of catastrophic acute axonal injury, degeneration of the distal segment of the injured axon creates a cellular and molecular environment that promotes effective axon regeneration (Simon and Watkins, 2018; Li et al., 2023); therefore, inhibiting SARM1 activation in human patients who experienced traumatic or ischemic nerve injury would probably not be beneficial, and might even be harmful.] Second, there should be a continued effort to discover additional SARM1 inhibitors that belong to different chemical classes and/or have different mechanisms of action. Finally, the best performing compounds should rapidly proceed to clinical trials. Given the incredible pace of research in this field over the last few years, it will probably not take too long before axon-protective medications become available for treatment of human patients with a broad array of peripheral – and possibly central – nervous system disorders. Disclosure statement The author receives research support from Astellas Gene Therapies (formerly known as Audentes Therapeutics, Inc) as a member of the muscle biopsy review committee for the ASPIRO (NCT03199469) and FORTIS (NCT04174105) clinical trials, which are evaluating the safety and efficacy of gene transfer therapy for X-linked myotubular myopathy (ASPIRO) and late onset Pompe disease (FORTIS). Acknowledgements I am grateful to Drs. Nigel G. Laing, Gina Ravenscroft, and Benedikt Schoser for helpful input during the conceptualization stage of this review. In addition, I would like to thank Ms. Christine Lin for assistance with figure preparation. References Abrams, R.M.C., Simpson, D.M., Navis, A., Jette, N., Zhou, L., and Shin, S.C. (2022). Small fiber neuropathy associated with SARS-CoV-2 infection. Muscle Nerve 65, 440-443. https://doi.org/10.1002/mus.27458 Bloom, A.J., Mao, X., Strickland, A., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2022). Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients. Mol Neurodegener 17, 1. https://doi.org/10.1186/s13024-021-00511-x Blume, G., Pestronk, A., Frank, B., and Johns, D.R. (1997). Polymyositis with cytochrome oxidase negative muscle fibres. Early quadriceps weakness and poor response to immunosuppressive therapy. Brain 120, 39-45. https://doi.org/10.1093/brain/120.1.39 Bosanac, T., Hughes, R.O., Engber, T., Devraj, R., Brearley, A., Danker, K., Young, K., Kopatz, J., Hermann, M., Berthemy, A., et al. (2021). Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 144, 3226-3238. https://doi.org/10.1093/brain/awab184 Bratkowski, M., Burdett, T.C., Danao, J., Wang, X., Mathur, P., Gu, W., Beckstead, J.A., Talreja, S., Yang, Y.S., Danko, G., et al. (2022). Uncompetitive, adduct-forming SARM1 inhibitors are neuroprotective in preclinical models of nerve injury and disease. Neuron 110, 3711-3726. https://doi.org/10.1016/j.neuron.2022.08.017 Britson, K.A., Ling, J.P., Braunstein, K.E., Montagne, J.M., Kastenschmidt, J.M., Wilson, A., Ikenaga, C., Tsao, W., Pinal-Fernandez, I., Russell, K.A., et al. (2022). Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci Transl Med 14, eabi9196. https://doi.org/10.1126/scitranslmed.abi9196 Conforti, L., Gilley, J., and Coleman, M.P. (2014). Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15, 394-409. https://doi.org/10.1038/nrn3680 Cummings, B.B., Marshall, J.L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A.R., Bolduc, V., Waddell, L.B., Sandaradura, S.A., O'Grady, G.L., et al. (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 9, eaal5209. https://doi.org/10.1126/scitranslmed.aal5209 De Reuck, J., De Coster, W., and vander Eecken, H. (1977). The target phenomenon in rat muscle following tenotomy and neurotomy. A comparative light microscopic and histochemical study. Acta Neuropathol 37, 49-53. https://doi.org/10.1007/BF00684540 Dingwall, C.B., Strickland, A., Yum, S.W., Yim, A.K., Zhu, J., Wang, P.L., Yamada, Y., Schmidt, R.E., Sasaki, Y., Bloom, A.J., et al. (2022). Macrophage depletion blocks congenital SARM1-dependent neuropathy. J Clin Invest 132, e159800. https://doi.org/10.1172/JCI159800 Engel, W.K. (1961). Muscle target fibres, a newly recognized sign of denervation. Nature 191, 389-390. https://doi.org/10.1038/191389a0 Erdmann, H., Scharf, F., Gehling, S., Benet-Pages, A., Jakubiczka, S., Becker, K., Seipelt, M., Kleefeld, F., Knop, K.C., Prott, E.C., et al. (2022). Methylation of the 4q35 D4Z4 repeat defines disease status in facioscapulohumeral muscular dystrophy. Brain, Epub ahead of print. https://doi.org/10.1093/brain/awac336 Feldman, H.C., Merlini, E., Guijas, C., DeMeester, K.E., Njomen, E., Kozina, E.M., Yokoyama, M., Vinogradova, E., Reardon, H.T., Melillo, B., et al. (2022). Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc Natl Acad Sci U S A 119, e2208457119. https://doi.org/10.1073/pnas.2208457119 Figley, M.D., Gu, W., Nanson, J.D., Shi, Y., Sasaki, Y., Cunnea, K., Malde, A.K., Jia, X., Luo, Z., Saikot, F.K., et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron 109, 1118-1136. https://doi.org/10.1016/j.neuron.2021.02.009 Gilley, J., Jackson, O., Pipis, M., Estiar, M.A., Al-Chalabi, A., Danzi, M.C., van Eijk, K.R., Goutman, S.A., Harms, M.B., Houlden, H., et al. (2021). Enrichment of SARM1 alleles encoding variants with constitutively hyperactive NADase in patients with ALS and other motor nerve disorders. Elife 10, e70905. https://doi.org/10.7554/eLife.70905 Goyal, N.A., Coulis, G., Duarte, J., Farahat, P.K., Mannaa, A.H., Cauchii, J., Irani, T., Araujo, N., Wang, L., Wencel, M., et al. (2022). Immunophenotyping of inclusion body myositis blood T and NK cells. Neurology 98, e1374-e1383. https://doi.org/10.1212/WNL.0000000000200013 Greenberg, S.A., Pinkus, J.L., Kong, S.W., Baecher-Allan, C., Amato, A.A., and Dorfman, D.M. (2019). Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 142, 2590-2604. https://doi.org/10.1093/brain/awz207 Griffin, J.W., Li, C.Y., Macko, C., Ho, T.W., Hsieh, S.T., Xue, P., Wang, F.A., Cornblath, D.R., McKhann, G.M., and Asbury, A.K. (1996). Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barre syndrome. J Neurocytol 25, 33-51. https://doi.org/10.1007/BF02284784 Hafer-Macko, C., Hsieh, S.T., Li, C.Y., Ho, T.W., Sheikh, K., Cornblath, D.R., McKhann, G.M., Asbury, A.K., and Griffin, J.W. (1996a). Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol 40, 635-644. https://doi.org/10.1002/ana.410400414 Hafer-Macko, C.E., Sheikh, K.A., Li, C.Y., Ho, T.W., Cornblath, D.R., McKhann, G.M., Asbury, A.K., and Griffin, J.W. (1996b). Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol 39, 625-635. https://doi.org/10.1002/ana.410390512 Hedberg-Oldfors, C., Lindgren, U., Visuttijai, K., Loof, D., Roos, S., Thomsen, C., and Oldfors, A. (2022). Respiratory chain dysfunction in perifascicular muscle fibres in patients with dermatomyositis is associated with mitochondrial DNA depletion. Neuropathol Appl Neurobiol 48, e12841. https://doi.org/10.1111/nan.12841 Hejbøl, E.K., Harbo, T., Agergaard, J., Madsen, L.B., Pedersen, T.H., Ostergaard, L.J., Andersen, H., Schroder, H.D., and Tankisi, H. (2022). Myopathy as a cause of fatigue in long-term post-COVID-19 symptoms: Evidence of skeletal muscle histopathology. Eur J Neurol 29, 2832-2841. https://doi.org/10.1111/ene.15435 Hiniker, A., Daniels, B.H., Lee, H.S., and Margeta, M. (2013). Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 1, 29. https://doi.org/10.1186/2051-5960-1-29 Hughes, R.O., Bosanac, T., Mao, X., Engber, T.M., DiAntonio, A., Milbrandt, J., Devraj, R., and Krauss, R. (2021). Small molecule SARM1 inhibitors recapitulate the SARM1(-/-) phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep 34, 108588. https://doi.org/10.1016/j.celrep.2020.108588 Huppke, P., Wegener, E., Gilley, J., Angeletti, C., Kurth, I., Drenth, J.P.H., Stadelmann, C., Barrantes-Freer, A., Bruck, W., Thiele, H., et al. (2019). Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp Neurol 320, 112958. https://doi.org/10.1016/j.expneurol.2019.112958 Inoue, M., Noguchi, S., Inoue, Y.U., Iida, A., Ogawa, M., Bengoechea, R., Pittman, S.K., Hayashi, S., Watanabe, K., Hosoi, Y., et al. (2023). Distinctive chaperonopathy in skeletal muscle associated with the dominant variant in DNAJB4. Acta Neuropathol 145, 235-255. https://doi.org/10.1007/s00401-022-02530-4 Kleefeld, F., Uruha, A., Schanzer, A., Nishimura, A., Roos, A., Schneider, U., Goebel, H.H., Schuelke, M., Hahn, K., Preusse, C., et al. (2022). Morphologic and molecular patterns of polymyositis with mitochondrial pathology and inclusion body myositis. Neurology 99, e2212-e2222. https://doi.org/10.1212/WNL.0000000000201103 Knauss, S., Preusse, C., Allenbach, Y., Leonard-Louis, S., Touat, M., Fischer, N., Radbruch, H., Mothes, R., Matyash, V., Bohmerle, W., et al. (2019). PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol Neuroimmunol Neuroinflamm 6, e558. https://doi.org/10.1212/NXI.0000000000000558 Korb, M.K., Kimonis, V.E., and Mozaffar, T. (2021). Multisystem proteinopathy: Where myopathy and motor neuron disease converge. Muscle Nerve 63, 442-454. https://doi.org/10.1002/mus.27097 Krause, K., Eggers, B., Uszkoreit, J., Eulitz, S., Rehmann, R., Guttsches, A.K., Schreiner, A., van der Ven, P.F.M., Furst, D.O., Marcus, K., et al. (2022). Target formation in muscle fibres indicates reinnervation - A proteomic study in muscle samples from peripheral neuropathies. Neuropathol Appl Neurobiol, e12853. https://doi.org/10.1111/nan.12853 Kurashige, T., Morino, H., Murao, T., Izumi, Y., Sugiura, T., Kuraoka, K., Kawakami, H., Torii, T., and Maruyama, H. (2022). TDP-43 accumulation within intramuscular nerve bundles of patients with amyotrophic lateral sclerosis. JAMA Neurol 79, 693-701. https://doi.org/10.1001/jamaneurol.2022.1113 Levine, T.D., and Pestronk, A. (1998). Inflammatory myopathy with cytochrome oxidase negative muscle fibers: methotrexate treatment. Muscle Nerve 21, 1724-1728. https://doi.org/10.1002/(sici)1097-4598(199812)21:12<1724::aid-mus15>3.0.co;2-2 Li, X., Zhang, T., Li, C., Xu, W., Guan, Y., Li, X., Cheng, H., Chen, S., Yang, B., Liu, Y., et al. (2023). Electrical stimulation accelerates Wallerian degeneration and promotes nerve regeneration after sciatic nerve injury. Glia 71, 758-774. https://doi.org/10.1002/glia.24309 Lukacs, M., Gilley, J., Zhu, Y., Orsomando, G., Angeletti, C., Liu, J., Yang, X., Park, J., Hopkin, R.J., Coleman, M.P., et al. (2019). Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Exp Neurol 320, 112961. https://doi.org/10.1016/j.expneurol.2019.112961 Margeta, M. (2020). Top ten discoveries of the year: Neuromuscular disease. Free Neuropathol 1, 4. https://doi.org/10.17879/freeneuropathology-2020-2627 Margeta, M. (2021). Neuromuscular disease: 2021 update. Free Neuropathol 2, 3. https://doi.org/10.17879/freeneuropathology-2021-3236 Margeta, M. (2022). Neuromuscular disease: 2022 update. Free Neuropathol 3, 5. https://doi.org/10.17879/freeneuropathology-2022-3805 Martin-Aguilar, L., Lleixa, C., and Pascual-Goni, E. (2022). Autoimmune nodopathies, an emerging diagnostic category. Curr Opin Neurol 35, 579-585. https://doi.org/10.1097/WCO.0000000000001107 McGonigal, R., Campbell, C.I., Barrie, J.A., Yao, D., Cunningham, M.E., Crawford, C.L., Rinaldi, S., Rowan, E.G., and Willison, H.J. (2022). Schwann cell nodal membrane disruption triggers bystander axonal degeneration in a Guillain-Barre syndrome mouse model. J Clin Invest 132, e158524. https://doi.org/10.1172/JCI158524 Mocciaro, E., Runfola, V., Ghezzi, P., Pannese, M., and Gabellini, D. (2021). DUX4 role in normal physiology and in FSHD muscular dystrophy. Cells 10, 3322. https://doi.org/10.3390/cells10123322 Oaklander, A.L., Mills, A.J., Kelley, M., Toran, L.S., Smith, B., Dalakas, M.C., and Nath, A. (2022). Peripheral neuropathy evaluations of patients with prolonged long COVID. Neurol Neuroimmunol Neuroinflamm 9, e1146. https://doi.org/10.1212/NXI.0000000000001146 Osterloh, J.M., Yang, J., Rooney, T.M., Fox, A.N., Adalbert, R., Powell, E.H., Sheehan, A.E., Avery, M.A., Hackett, R., Logan, M.A., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481-484. https://doi.org/10.1126/science.1223899 Riva, N., Gentile, F., Cerri, F., Gallia, F., Podini, P., Dina, G., Falzone, Y.M., Fazio, R., Lunetta, C., Calvo, A., et al. (2022). Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain 145, 276-284. https://doi.org/10.1093/brain/awab285 Sarparanta, J., Jonson, P.H., Kawan, S., and Udd, B. (2020). Neuromuscular diseases due to chaperone mutations: A review and some new results. Int J Mol Sci 21, 1409. https://doi.org/10.3390/ijms21041409 Schiaffino, S., Rossi, A.C., Smerdu, V., Leinwand, L.A., and Reggiani, C. (2015). Developmental myosins: expression patterns and functional significance. Skelet Muscle 5, 22. https://doi.org/10.1186/s13395-015-0046-6 Sewry, C.A., Feng, L., Chambers, D., Matthews, E., and Phadke, R. (2021). Importance of immunohistochemical evaluation of developmentally regulated myosin heavy chains in human muscle biopsies. Neuromuscul Disord 31, 371-384. https://doi.org/10.1016/j.nmd.2021.02.007 Shi, Y., Kerry, P.S., Nanson, J.D., Bosanac, T., Sasaki, Y., Krauss, R., Saikot, F.K., Adams, S.E., Mosaiab, T., Masic, V., et al. (2022). Structural basis of SARM1 activation, substrate recognition, and inhibition by small molecules. Mol Cell 82, 1643-1659. https://doi.org/10.1016/j.molcel.2022.03.007 Simon, D.J., and Watkins, T.A. (2018). Therapeutic opportunities and pitfalls in the treatment of axon degeneration. Curr Opin Neurol 31, 693-701. https://doi.org/10.1097/WCO.0000000000000621 Stevanovski, I., Chintalaphani, S.R., Gamaarachchi, H., Ferguson, J.M., Pineda, S.S., Scriba, C.K., Tchan, M., Fung, V., Ng, K., Cortese, A., et al. (2022). Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci Adv 8, eabm5386. https://doi.org/10.1126/sciadv.abm5386 Vallat, J.M., Magy, L., Corcia, P., Boulesteix, J.M., Uncini, A., and Mathis, S. (2020). Ultrastructural lesions of nodo-paranodopathies in peripheral neuropathies. J Neuropathol Exp Neurol 79, 247-255. https://doi.org/10.1093/jnen/nlz134 Volpatti, J.R., Ghahramani-Seno, M.M., Mansat, M., Sabha, N., Sarikaya, E., Goodman, S.J., Chater-Diehl, E., Celik, A., Pannia, E., Froment, C., et al. (2022). X-linked myotubular myopathy is associated with epigenetic alterations and is ameliorated by HDAC inhibition. Acta Neuropathol 144, 537-563. https://doi.org/10.1007/s00401-022-02468-7 Wang, X., Jia, Y., Zhao, J., Lesner, N.P., Menezes, C.J., Shelton, S.D., Venigalla, S.S.K., Xu, J., Cai, C., and Mishra, P. (2022). A mitofusin 2/HIF1alpha axis sets a maturation checkpoint in regenerating skeletal muscle. J Clin Invest 132, e161638. https://doi.org/10.1172/JCI161638 Weihl, C.C., Topf, A., Bengoechea, R., Duff, J., Charlton, R., Garcia, S.K., Dominguez-Gonzalez, C., Alsaman, A., Hernandez-Lain, A., Franco, L.V., et al. (2023). Loss of function variants in DNAJB4 cause a myopathy with early respiratory failure. Acta Neuropathol 145, 127-143. https://doi.org/10.1007/s00401-022-02510-8 Winkler, M., von Landenberg, C., Kappes-Horn, K., Neudecker, S., Kornblum, C., and Reimann, J. (2021). Diagnosis and clinical development of sporadic inclusion body myositis and polymyositis with mitochondrial pathology: A single-center retrospective analysis. J Neuropathol Exp Neurol 80, 1060-1067. https://doi.org/10.1093/jnen/nlab101
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |