|
|
|
Free Neuropathology 3:11 (2022) |
|
Letter |
|
A potential diagnostic pitfall: Primary synovial sarcoma of the central nervous system |
|
Arnault Tauziède-Espariat1,2, Nicolas Macagno3, Daniel Pissaloux3, Dominique Figarella-Branger4,5, Romain Appay4,5, Dorian Bochaton6, Sanaa Tazi7, Paul Kauv8, Lauren Hasty1, Alice Métais1,2, Fabrice Chrétien1, Pascale Varlet1,2 |
|
1 Department of Neuropathology, GHU Paris - Psychiatry and Neuroscience, Sainte-Anne Hospital, Paris, France |
|
Corresponding author: |
|
Submitted: 14 March 2022 Accepted: 25 April 2022 Copyedited by: Cinthya Agüero Published: 26 April 2022 |
|
Keywords: SS18, SSX, Synovial sarcoma, BCOR |
|
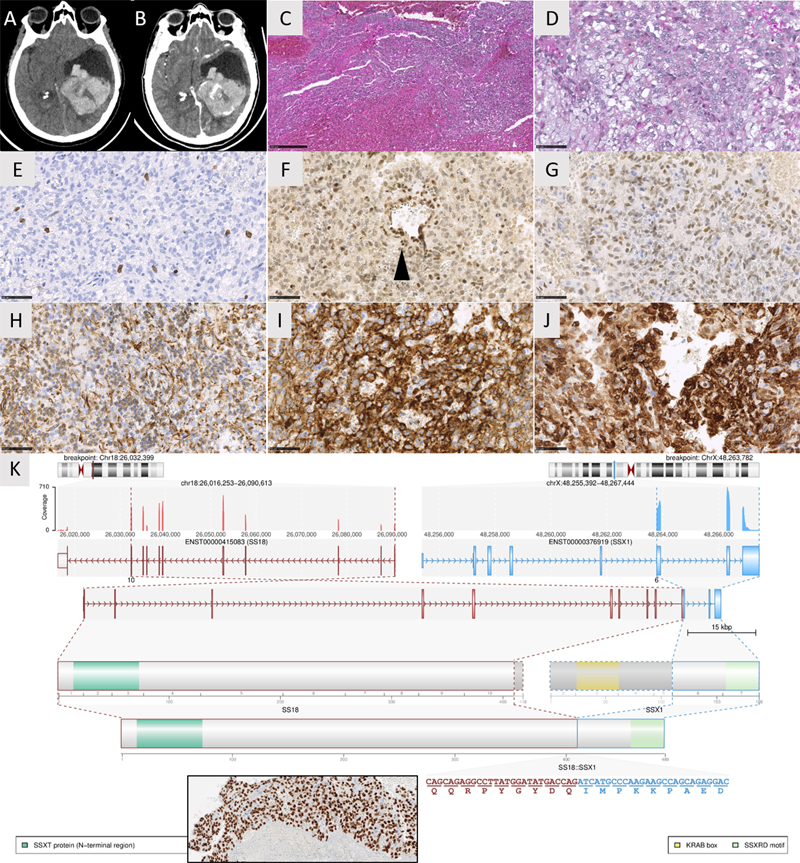
Introduction Synovial sarcoma (SS) is an aggressive soft tissue sarcoma that occurs primarily in the juxta-articular extremital regions of young adults, followed by the trunk and the head and neck. They are universally defined by a fusion of SS18 with one of the SSX genes (predominantly SSX1). Primary SS of the central nervous system (CNS) have been rarely reported in the literature (n=22 molecularly confirmed cases) and have not yet been added to the mesenchymal non-meningothelial chapter of the World Health Organization (WHO) classification of CNS tumors. Here, we report one case of SS18-rearranged CNS-SS, with a review of the literature to determine its clinical, radiological, and histopathological features and its main differential diagnoses. Case presentation A 62-year-old woman presented with an intraventricular hemorrhage (Figure 1 A-B). The patient had a history of a hemorrhagic lesion diagnosed as a central neurocytoma and was treated by surgery 3 years earlier. The tumor exhibited histologically dense, solid, hemorrhagic, and basophilic cell proliferation (Figure 1C), composed of monotonous spindle cells, with round to oval nuclei lacking any pleomorphism or mitotic activity. The cytoplasm was scant or more abundant and vacuolated (Figure 1D). There was no collagenous stroma, myxoid changes, “hemangiopericytoma-like” vasculature, whorls, psammoma bodies, neurocytic rosettes, papillae, rhabdoid, or glandular formations. Necrosis and microvascular proliferation were absent, and the Ki67 labeling index was low (1%) (Figure 1E). Tumor cells were immunonegative for AE1/AE3, CK7, CK20, CK18, SSTR2A, EMA, CD34, STAT6, CD45, CD99, NUT, Olig2, GFAP, SOX10, PS100, HMB45, chromogranin A, synaptophysin, NeuN, neurofilament, desmin, and smooth muscle actin. The expression of H3K27me3 and BRG1 was retained. INI1 was maintained with a diminished expression in comparison to the endothelium (Figure 1F). The tumor expressed vimentin, CD56, Bcl2, TLE1, and BCOR (but with less intense staining compared to our control, a molecularly confirmed CNS tumor with internal tandem duplication of BCOR) (Figure 1G-J). Histopathological characteristics were like those of the previous surgical sample. The initial tumor was diagnosed as a neurocytoma due to its intraventricular location, its expression of CD56 and because of its absence of any sign of malignancy (particularly no mitosis). Initial and recent whole-body imaging did not show other tumor locations. Digital droplet PCR failed to reveal an internal tandem duplication of BCOR, and whole-exome RNA sequencing [1] demonstrated a SS18::SSX1 gene fusion (Figure 1K). An additional immunostaining targeting the chimeric protein SS18::SSX was secondarily performed and revealed a strong and diffuse expression in the primary tumor and its recurrence (Figure 1K). Using DNA-methylation analysis, the tumor was not classifiable by the Heidelberg Brain Tumor Classifier (v12.5) and the Sarcoma Classifier (v12.2). A final diagnosis of primary CNS-SS was suggested. Forty months after the initial diagnosis, the patient was alive without a tumor residue.
Figure 1. Radiological, histological, and molecular features of the recurrent tumor. (A) Axial CT scan showing a voluminous left intraventricular mass with hemorrhage. (B) Axial CT scan after contrast injection showing a heterogeneous enhancement of the mass. (C) Densely cellular proliferation with hemorrhagic changes (HPS, magnification x100). (D) Monotonous basophilic spindle cells with round to oval nuclei and vacuolated cytoplasm (HPS, magnification x400). (E) Low Ki67 labeling index (magnification x400). (F) retained expression of INI1 with low intensity (magnification x400). (G) Diffuse but moderate immunoexpression of BCOR (magnification x400). (H) Diffuse immunoexpression of Vimentin (magnification x400). (I) Diffuse immunoexpression of CD56 (magnification x400). (J) Diffuse immunoexpression of Bcl2 (magnification x400). (K) Whole exome RNA-seq analysis highlights a fusion between SS18 (pink) and SSX1 (blue) genes, respectively located on chr.18 and chrX. Immunopositivity for the chimeric protein between SS18 and SSX with retained protein domain of SS18 detected by immunohistochemistry (insert magnification x400). Black scale bars represent 50 μm for figures D-J; and 250 μm for figure C. HPS: Hematoxylin Phloxin Saffron. Clicking the figure will lead you to a virtual slide (H&E). Discussion and conclusion Molecularly confirmed examples of primary CNS-SS are very rarely reported in the literature (n=22 cases, Table S1) [2–8]. Like their soft tissue counterpart, they affect young adults (median age 21 years with ranges from 1 to 81) or children (32% of cases) [2,3] with a male predominance (sex ratio male to female: 1.75) [2–8]. They are mainly supratentorial (19/22, 86% of cases) [2–8] and 9% of them are intraventricular as was the case for our patient [2]. Clinical symptoms depend on the location of the tumor, but interestingly, 4/22 cases (18%) are revealed by an intracerebral hematoma [2,5,8]. Radiologically, they present as a solid (13/22 cases) or cystic mass (8/22 cases) that is well defined from the parenchyma (11/16 cases) [2–4,8]. Some of them present a dural attachment (4/22 cases) [4–7]. Histopathologically, SS is classically divided into three subtypes within the soft tissue: biphasic (composed of a mixture of spindle cells and epithelial component), monophasic (only composed of spindle cells), and poorly differentiated (composed of round or spindle cells with a high mitotic index and pronounced atypia). Rare SS examples can exhibit a round cell morphology [9–11]. In the CNS, all these variants have been reported, the monophasic subtype (as our case) being the most frequent (10/21 cases compared to 9/21 biphasic and 2/21 poorly differentiated cases) [2,4–7]. Recently, a highly specific and sensitive antibody targeting the SS18::SSX fusion protein has been developed independently of the SS18 fusion gene partner among SSX genes [12], which can serve as a surrogate to molecular testing [12,13]. Like their soft tissue counterparts, SS frequently and strongly express Bcl2, vimentin, and CD99, which are not specific. The classical phenotype includes a variable, usually focal aberrant epithelial phenotype, diffuse and intense nuclear expression of TLE1 [14], and reduced expression of INI1 [15]. The glial markers, SSTR2A, CD34, and STAT6, are not expressed, which allows one to rule out differential diagnoses (gliosarcoma, meningioma, and solitary fibrous tumor). However, the occasional expression of BCOR may be a confusing finding [16], but additional molecular analyses did not confirm the presence of BCOR alterations and found a SS18::SSX1 fusion, the most frequent fusion transcript described in soft tissue SS. In the CNS, no detailed fusion has been reported, except in one case (SS18::SSX2) because only FISH analyses for SS18 have been performed [2–8]. Like many fusion-driven neoplasms, the cellular origin of SS remains unknown. A mesenchymal hypothesis was suggested with respect to its preferential location in soft tissue. Because meningeal involvement has been reported in a part of CNS cases, it is reasonable to suggest a possible mesenchymal dural origin. Recently, DNA-methylation-based classification of CNS tumors has been shown to be a robust tool for molecular tumor classification, most likely because individual tumor types maintain an epigenetic ‘memory’ of their distinct cell of origin [17]. In v12.2 of the sarcoma classifier, there is a methylation class of synovial sarcoma [18]. However, due to the low cellularity in our hemorrhagic sample, the DNA methylation profiling did not properly cluster and did not allow us to classify this tumor. The prognosis of soft tissue SS depends on the FNCLCC grade [19] and genomic index [20]. CNS-SS is associated with a unfavorable outcome, high rates of recurrence (18/21 cases, 86%), and death (16/21 cases, 76%) with a median overall survival of 14 months [2–5,7,8]. Our case represents an exception with the longest overall survival ever described without adjuvant treatment (40 months), possibly due to the low histological grade of the tumor. In conclusion, CNS SS with a SS18::SSX1 fusion represents a rare mesenchymal non-meningothelial tumor, not yet included in the WHO classification of CNS tumors. Although its diagnosis remains challenging, different diagnostic tools (immunohistochemistry, FISH, RNA sequencing) exist to identify SS. Further studies are needed to elucidate its cellular origin in the CNS. Declarations Ethics approval This study was approved by the GHU Paris Psychiatry Neurosciences, Sainte-Anne Hospital’s local ethic committee. Consent for publication The patient signed informed consent forms. Competing interests The authors declare that they have no conflicts of interest directly related to the topic of this article. Funding The authors declare that they have not received any funding. Authors’ contributions ATE, PK and ST compiled the MRI and clinical records; ATE, AM, FC and PV conducted the neuropathological examinations; NC, DP, DFB, RA and DB conducted the molecular studies; ATE, LH, and PV drafted the manuscript. All authors reviewed the manuscript. Acknowledgements We would like to thank the laboratory technicians at the GHU Paris Neuro Sainte-Anne for their assistance. References
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |