|
Abstract
Alzheimer disease is one of the most challenging demons in our society due to its very high prevalence and its clinical manifestations which cause deterioration of cognition, intelligence, and emotions – the very capacities that distinguish Homo sapiens from other animal species. Besides the personal, social, and economical costs, late stages of AD are vivid experiences for the family, relatives, friends, and general observers of the progressive ruin of an individual who turns into a being with lower mental and physical capacities than less evolved species. A human brain with healthy cognition, conscience, and emotions can succeed in dealing with most difficulties that life may pose. Without these capacities, the same person probably cannot. Due, in part, to this emotional impact, the absorbing study of AD has generated, over the years, a fascinating and complex story of theories, hypotheses, controversies, fashion swings, and passionate clashes, together with tremendous efforts and achievements geared to improve understanding of the pathogenesis and treatment of the disorder. Familal AD is rare and linked to altered genetic information associated with three genes. Sporadic AD (sAD) is much more common and multifactorial. A major point of clinical discussion has been, and still is, establishing the differences between brain aging and sAD. This is not a trivial question, as the neuropathological and molecular characteristics of normal brain aging and the first appearance of early stages of sAD-related pathology are not easily distinguishable in most individuals. Another important point is confidence in assigning responsibility for the beginning of sAD to a few triggering molecules, without considering the wide number of alterations that converge in the pathogenesis of aging and sAD. Genetic risk factors covering multiple molecular signals are increasing in number. In the same line, molecular pathways are altered at early stages of sAD pathology, currently grouped under the aegis of normal brain aging, only to increase massively at advanced stages of the process. Sporadic AD is here considered an inherent, natural part of human brain aging, which is prevalent in all humans, and variably present or not in a few individuals in other species. The progression of the process has devastating effects in a relatively low percentage of human beings eventually evolving to dementia. The continuum of brain aging and sAD implies the search for a different approach in the study of human brain aging at the first stages of the biological process, and advances in the use of new technologies aimed at slowing down the molecular defects underlying human brain aging and sAD at the outset, and transfering information and tasks to AI and coordinated devices.
Index
Summary
1. Introduction
2. β-amyloid and Tau
2a. β-amyloid (Aβ)
2b. Tau
3. Familial AD (fAD; early-onset familial Alzheimer’s disease: EOFAD), and the β-amyloid cascade hypothesis
4. Sporadic AD (sAD; Late-onset Alzheimer disease: LOAD)
5. NFTs and SPs in non-human brain aging
6. Synapses
7. Neurotransmitters, neuromodulators, and related receptors
7a. Acetylcholine (Ach) and acetylcholine receptors (AChR)
7b. Glutamate and glutamate receptors (GluRs)
7c. γ-aminobutyric acid (GABA) and GABA receptors
7d. Serotonin and 5-hydroxytryptamine (5-HT) receptors
7e. Noradrenergic system
7f. Adenosine receptors
7g. Endocannabinoids and cannabinoid receptors (CBRs)
8. Trophic factors and receptors
9. Endoplasmic reticulum stress
10. Failure to remove debris: the ubiquitin-proteasome system (UPS) and autophagy in sAD
11. Granulovacuolar degeneration (GVD)
12. Glial alterations in aging and sAD
12a. Astrocytes
12b. Microglia
12c. Oligodendrocytes
13. The neurovascular system in AD
14. Purine and pyrimidine metabolism in sAD
15. Epigenetics in brain aging and sAD
15a. Histone modifications, DNA methylation, and hydroxymethylation
15b. Non-coding RNAs
16. Microorganisms and sAD
16a. Microorganisms in the brain and oral cavity
16b. Gut microbiota
17. Seeding and spreading of β-amyloid and tau
17a. Seeding β-amyloid
17b. Tau seeding
17c. Multiple seeding foci of β-amyloid and tau pathology; vulnerable and resistant populations to tau seeding in brain aging and sAD
18. Neuronal death
19. Neuronal connectivity networks in brain aging and sAD
20. Human brain aging and preclinical AD
21. Primary age-related tauopathy (PART), rapidly progressive sAD, and sAD resilience
21a. PART
21b. Rapidly progressive AD
21c. sAD resilience
22. Biochemical changes beyond tau and β-amyloid at the the first stages of NFT pathology
22a. Aberrant cell-cycle re-entry, and altered adult neurogenesis
22b. Brain lipids
22c. Lipid rafts and cell membranes
22d. Mitochondria
22e. Oxidative stress damage
22f. Inflammation
22g. Protein synthesis impairment
22h. Dysregulated protein phosphorylation
23. Concluding comments
Abbreviations
Funding
Acknowledgements
References
Summary
This is a comprehensive historical and up-dated review on the pathogenesis of Alzheimer’s disease (AD) in relation to intrinsic process of natural brain aging. The study covers, in addition to β-amyloid and tau pathology, alterations in multiple merging molecular pathways and sub-cellular structures underpinning brain aging and AD.
Familial AD (fAD) is rare and linked to altered genetic information associated with three genes. Sporadic AD (sAD) is much more common and multifactorial. A major point of clinical discussion has been, and remains, establishing the differences between brain aging and sAD. This is not a trivial question, as the neuropathological and molecular characteristics of normal brain aging and the first appearance of early stages of sAD-related pathology are not easily distinguishable in most individuals. Another important point is confidence in assigning responsibility for the beginning of sAD to a few triggering molecules, without considering the wide number of alterations that converge in the pathogenesis of aging and sAD. Recognized genetic risk factors covering multiple molecular signals are increasing in number. Molecular alterations of lipid rafts, protein synthesis from the nucleolus to the ribosome, protein phosphorylation, kinase activation, purine metabolism, epigenetic regulation of DNA and RNA, mitochondria and energy metabolism, inflammation, oxidative stress, cell-cycle re-entry, and cell death precede, in some regions (i.e., frontal cortex), abnormal tau deposition and amyloid plaques. Human brain aging and sAD do not follow a linear logic based on the assumption that a cause results in one or several effects; several separate alterations converge and potentiate each other to incorporate anomalies in additional pathways. Tau seeding and spreading are active intercellular and intracellular processes that explain, only in part, disease progression. Cell and region vulnerability are essential elements. Brain aging with neurofibrillary tangles (NFTs) restricted to the temporal lobe and selected nuclei of the brain stem, primary age-related tauopathy, preclinical AD, mild cognitive impairment (MCI) of Alzheimer type, typical AD, rapid progressive AD, and AD subtypes, are forms of sAD modulated by individual genetic and molecular factors. As in atherosclerosis, the progression of the process has devastating effects in a relatively low percentage of human beings. Future modulation of human brain aging and sAD will require the combined application of Artificial Intelligence, brain DNA editing, external electrical or wave-based signals to reduce energy consumption, and optimization of mitochondrial function, together with implantation of microdevices, to facilitate cooperative human-machine operation, pharmacological protection of lipid-protein interactions, high-throughput molecular technology, and resetting during sleep stages.
1. Introduction
The clinical and neuropathological characteristics and clinical correlates of Alzheimer disease (AD) have been described in several recent reviews (1-9). However, the study of AD has generated a fascinating and complex compendium of theories, hypotheses, controversies, fashion swings, and passionate clashes, together with tremendous efforts and achievements geared to improve understanding of the pathogenesis and treatment of the disorder. The present paper is a critical review of brain aging and AD that includes molecular abnormalities and early metabolic alterations beyond β-amyloid and tau pathology. These changes, together with genetic factors, converge in the pathogenesis of AD. Learning about early molecular modifications preceding by many years the appearance of clinical symptoms, when present, will serve to improve understanding of brain aging and the AD continuum.
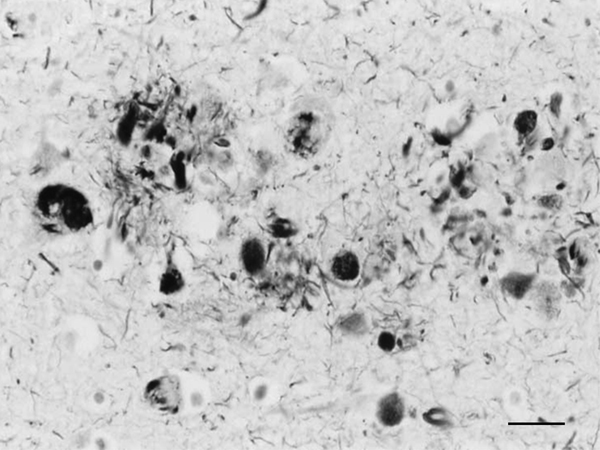
Figure 1: Dystrophic neurites of SPs and NFTs in the frontal cortex of a 76-year-old woman with dementia. Paraffin section, Gros-Bielschowsky silver method without counterstaining, black and white figure, bar = 25μm.
Until the beginning of the last century, cognitive impairment and dementia were considered natural features of old age. Multiple brain infarcts were common in old people, and vascular dementia due to arteriopathy was thought to be the main cause of senile dementia. However, microscopic study of post-mortem brains stained with the dyes available at that time revealed the presence of certain structural anomalies in aged individuals. Paul Block and Georges Marinesco (10) described “amas ronds”, and Emil Redlich (11) “miliare Sklerose” in the neuropil, interpreted at that time as nodules of glial sclerosis, which we now know as senile plaques (SPs). The introduction of the Max Bielschowsky silver method allowed visualisation of argyrophilic structures in neurons. Using this method, Alois Alzheimer described for the first time large numbers of argyrophilic neurofibrillary tangles (NFTs) and aggregates of dystrophic neurites in the brain of a 51-year-old woman who had suffered from progressive dementia and hallucinations in the previous four and half years (12). Other cases were published shortly afterwards (13). The term Alzheimer’s pre-senile dementia was introduced by Emil Kraepelin (14) to define the combination of pre-senile (before the age of 65) dementia in individuals with the morphological lesions described by Alzheimer. Oskar Fischer (15), using the same method, described the presence of ‘Drusen’ or ‘drusige Nekrosen’ in 16 cases of senile dementia characterized by loss of memory and sense of location, disorientation, and confabulation. Subsequent Fischer reports (16, 17) detailed the morphology of abnormal fibrils and abnormal neurites, and their stages of formation, in a large series of older individuals. The term ‘‘senile plaque’’ (SP) for these structures was proposed by Simchowitz (18). Fischer also described “drusige Entartung der Gefässe” which corresponds to amyloid angiopathy. Interestingly, Fischer also reported and illustrated the presence of NFTs in the same cases with dementia (19). Hundreds of articles appeared in the succeeding years. Alzheimer focused on NFTs as the main cause of dementia, whereas Fischer thought that SPs were the main substrate of dementia in older cases. Moreover, Alzheimer contemplated NFTs as aggregates of abnormal neurofibrils, while Fischer considered dystrophic neurites of SPs composed of abnormal neurofibrils, and NFTs a particular abnormality of nerve cells (19). Bielschowsky proposed a link between tangles and neuritic changes (20) (Figures 1 and 2). NFTs and SPs are now considered AD-related pathology or AD-neuropathologic change (ADNC) (https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf). The term Pick’s disease (PiD) was coined in 1926 to distinguish AD from PiD primary frontotemporal degenerative atrophy (21). As late as the 1960s, AD and PiD were considered early dementias, whereas pure senile dementia, vascular dementias, and mixed (vascular and degenerative) were classified as dementias in old age (22). The frontiers between AD and pure senile dementia were not clear, as the onset of clinical symptoms in many cases classified as AD was after the age of sixty (23). It was not until the 1970s that Alzheimer’s pre-senile dementia and senile dementia with changes of Alzheimer type were considered to be within the same spectrum (24-27). The inclusive term “Alzheimer-Fischer dementia” was never contemplated.
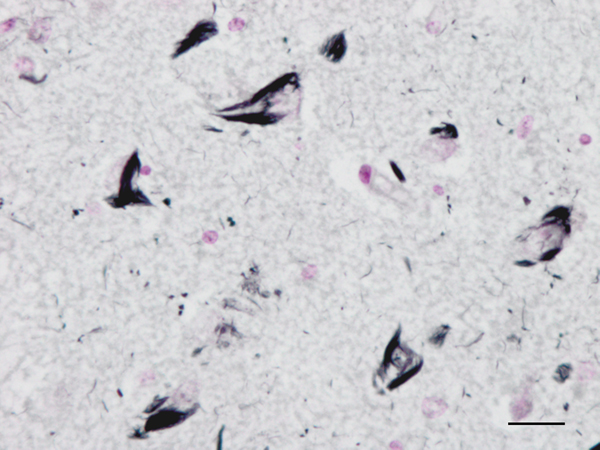
Figure 2: Neurofibrillary tangles in the CA1 region of the hippocampus of a man aged 69 years with no apparent cognitive impairment. Paraffin section, Gallyas staining, lightly counterstained with haemtoxylin, bar = 25μm.
The first approach toward a clinical consensus on AD was made in 1984; clinical diagnosis of AD was set up in three categories – possible, probable, and definite (requiring neuropathological verification) (28). Definite AD was fixed as a neurodegenerative disease manifested by progressive dementia with a neuropathological substrate characterized by brain atrophy, neuronal death, and a particular distribution of abundant SPs and NFTs in the brain.
Box 1: Clinical classification of Alzheimer’s disease (https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf).
Preclinical Alzheimer’s disease
In this phase, individuals may have measurable brain changes that indicate the earliest signs of AD (biomarkers), but they have not yet developed symptoms such as memory loss.
Mild cognitive impairment due to Alzheimer’s disease
People with MCI due to AD have biomarker evidence of Alzheimer’s brain changes plus new but subtle symptoms such as problems with memory, language and thinking. These cognitive problems may be noticeable to the individual, family members and friends, but not to others, and they may not interfere with individuals’ ability to carry out everyday activities.
Mild Alzheimer’s dementia
In the mild stage of Alzheimer’s dementia, most people are able to function independently in many areas but are likely to require assistance with some activities to maximize independence and remain safe. Handling money and paying bills may be especially challenging, and they may need more time to complete common daily tasks. They may still be able to drive, work and participate in their favorite activities.
Moderate Alzheimer’s dementia
In the moderate stage of Alzheimer’s dementia, which is often the longest stage, individuals experience more problems with memory and language, are more likely to become confused, and find it harder to complete multistep tasks such as bathing and dressing. They may become incontinent at times, and they may start having personality and behavioral changes, including suspiciousness and agitation. They may also begin to have problems recognizing loved ones.
Severe Alzheimer’s dementia
In the severe stage of Alzheimer’s dementia, individuals’ ability to communicate verbally is greatly diminished, and they are likely to require around-the-clock care. Because of damage to areas of the brain involved in movement, individuals become bed-bound. Being bed-bound makes them vulnerable to physical complications including blood clots, skin infections and sepsis, which triggers body-wide inflammation that can result in organ failure. Damage to areas of the brain that control swallowing makes it difficult to eat and drink. Because of this, food particles may be deposited in the lungs and cause lung infection.
In contrast to AD dementia, well-tolerated progressive slower processing, memory loss particularly related to recent events, more trouble multitasking, slight cognitive decline, sleep disorder, emotional changes, slight or moderate depression, and bilateral brain activation for memory functions developing around the sixties are all consistent with “normal brain aging”. Neuropathological alterations in normal old-aged individuals are NFTs in the hippocampus, entorhinal cortex, and inferior temporal cortex, and very rarely in the frontal neocortex; the distribution of SPs, if present, is more heterogeneous (29-35).
Clinical and neuropathological criteria to identify borderline cases between AD and cognitive impairment due to normal brain aging yielded only a limited consensus (36, 37). A few years later, CERAD proposed a neuritic plaque score based on the number of plaques per mm2 and the age of the individual to categorize AD in comparison to normal brain aging (38, 39).
Evidence of a clinical progression and post-mortem neuropathological observations showing a concatenation of AD-related changes in old age and sAD (29-32, 40-43) prompted a clinical redefinition of AD at the beginning of the second decade of this century.
A crucial approach was the combination of clinical criteria, biochemical biomarkers in body fluids, and neuroimaging techniques to define the diagnosis of preclinical AD, mild cognitive impairment (MCI) due to AD, and AD (44-50).
More precise clinical definitions have been proposed to categorize different stages of AD (51). The classification shown in Box 1 is a summarized transcription of the Alzheimer’s association report: 2022 Alzheimer’s disease facts and figures (https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf).
The American Academy of Neurology estimates that MCI is present in about 8% of people age 65 to 69, in 15% of 75- to 79-year-olds, in 25% of those age 80 to 84, and in about 37% of people 85 years of age and older. About 7.5% will develop dementia in the first year after diagnosis of MCI; about 15% will develop dementia in the second year; about one third will develop dementia due to AD within five years (52, 53). The prevalence of dementia in 65-69-year-olds is approximately 0.01% of individuals; the prevalence of dementia doubles with increments of five years; thereby, between 25% and 50% of individuals over the age of 85 suffer from dementia (2). It is estimated that between 50% and 80% of cases with dementia have AD (2). Age, gender, race, living conditions, and genetic factors mark differences in the duration of preclinical and dementia stages in sAD (54, 55).
2. β-amyloid and Tau
Electron microscopic studies revealed that NFTs were composed of paired helical filaments (PHFs) that disrupted the architecture of the cyto-skeleton. SPs were forged from a core of compact fibrils consistent with amyloids surrounded by dystrophic neurites filled with altered mitochondria, vesicles, numerous pleomorphic residual bodies, and PHFs (56-61) (Figure 3).
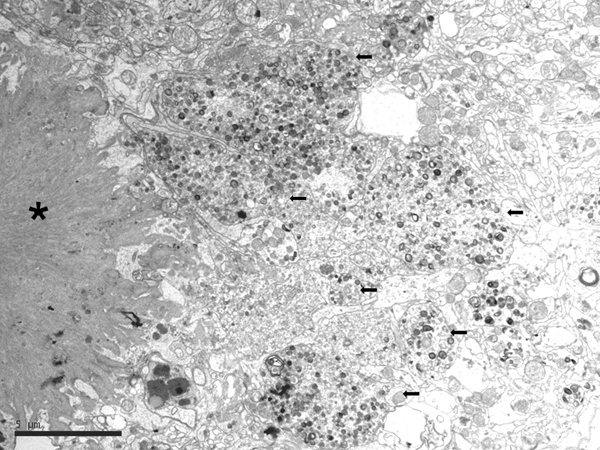
Figure 3: Electron microscopy of an SP showing the central core of amyloid fibrils (asterisk) and peripheral dystrophic neurites (black arrows) filled with vesicles, dense bodies, abnormal mitochondria, and paired helical filaments; bar = 5μm.
2a. β-amyloid (Aβ)
Subsequently, molecular studies identified β-amyloid as the main component of cerebral amyloid in β-amyloid angiopathy and SPs (62-66).
The amyloid precursor protein (APP) is a transmembrane protein which modulates brain cell adhesion, synaptic plasticity, and multiple intracellular signaling through the small endodomain of the molecule. APP processing is regulated by cytoplasmic phosphorylation (67). Cleavage of APP occurs through the combined action of α-, β-, and δ-secretases. β-secretase (BACE) is a GPI-anchored aspartyl protease (68). γ-secretase is a coprotein complex mainly composed of presenilin 1 (PSEN1) and presenilin 2 (PSEN2); components of the γ-secretase complex aph-1 homolog A; γ-secretase subunit (APH1A); APH1B; nicastrin (NCT/NCSTN); and presenilin enhancer γ-secretase subunit (PEN2/PSENEN), together with the modulators neprilysin (NEP/MME) and insulin-degrading enzyme (IDE). The γ-secretase complex is considered the “proteasome of the membrane” because of its capacity to act as a protelytic enzyme on more than 90 substrates (69-71). Cleavage of APP through α- and δ-secretase leads to the non-amyloidogenic pathway of APP degradation, whereas the combined action of β- and δ-secretases generates small truncated C-teminal peptides at positions 42 (Aβ1-42 or Aβ42) or 40 (Aβ1-40 or Aβ40), depending on the thickness of the membrane, and many other small forms are amyloidogenic as well (72-75). Local cholesterol content affects the various secretase activities (76), including cholesterol derived from astrocytes (77).
Low physiological concentrations of Aβ seem necessary for long-term potentiation induction and for memory formation, probably acting on cAMP and cGMP (78). However, in aging and AD there is not only abnormal production of β-amyloid. Aβ is aggregated and accumulates in the extracellular space due to its hydrofobicity, facility for oligomerization, and transformation from an α-helix to a β-sheet conformation (66, 73, 74). Several enzymes can degrade β-amyloid such as neprilysin, plasmin, endothelin-converting enzymes, angiotensin-converting enzymes, insulin-degrading enzyme, several matrix proteinases, and cathepsins A and B (79, 80). Soluble Aβ is drained across the lymphatic wall, binding to low-density lipoprotein receptor-related protein (LRP-1). The expression levels of some of these enzymes and transporters are reduced in AD (81-85). Impaired lymphatic drainage and altered blood vessel walls impair the elimination of soluble β-amyloid via the circulatory system (86, 87) (see section 13).
Astrocytes followed by neurons are the main source of clusterin in brain; clusterin is then released to the extracellular space (88). Clusterin expression is increased in AD (87, 88), and co-localizes with β-amyloid deposits (91), more specifically with Aβ1-40 (92). Clusterin may act as an extracellular chaperone (93) and it contributes to early stages of β-amyloid plaque pathology (94). In addition to being involved in Aβ aggregation and clearance and in the modulation of Aβ transport across the blood brain barrier (BBB) (95-97), clusterin is also known to reduce Aβ toxicity (98, 99). In addition, clusterin seems to interact with bridging integrator protein 1 (BIN1) and tau (100).
The main β-amyloid that circulates in brain interstitial fluid and cerebrospinal fluid (CSF) is soluble Aβ40. β-amyloid deposits in AD are categorized as primitive or immature plaques, mature or neuritic plaques (classical SPs), compact or burned-out, cotton-wool plaques, diffuse plaques, subpial β-amyloid deposits, β-amyloid angiopathy, and perivascular plaques (dyshoric angiopathy) (Figure 4). β-amyloid can also be found in the cytoplasm of neurons, and in astrocytes at the periphery of SPs. β-amyloid is composed of a mixture of peptides of different molecular weight: Aβ40 and Aβ42 are predominant in SPs, while Aβ40 is mainly located at the core of SPs and Aβ42 at their periphery. Diffuse plaques contain Aβ42 and truncated forms Aβ17-42. Subpial β-amyloid deposits are mainly composed of amino-terminal truncated species. β-amyloid species have different aggregation properties. N-terminal truncated Aβ with pyroglutamate modification at position 3 and Aβ phosphorylated at serine 8 show enhanced aggregation into oligomers and fibrils. These forms appear at late stages (biochemical stages 2 and 3) of β-amyloid formation, whereas soluble and insoluble aggregates composed of non-modified Aβ are found at early stages (stage 1) (101, 102).
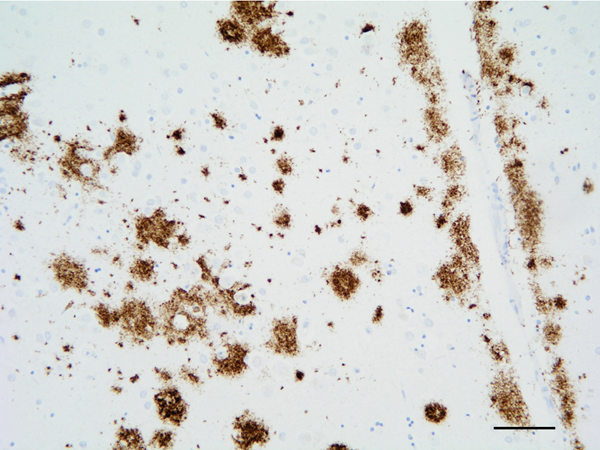
Figure 4: β-amyloid deposits in the temporal cortex. Paraffin section, β-amyloid immunohistochemistry, slight hematoxylin counterstaing, bar = 50μm.
Soluble β-amyloid oligomers (AβOs) and amyloid-β derived diffusible ligands (ADDLs), acting through specific cell surface receptors rather than fibrils, are toxic and cause neurodegeneration (103-115). High-molecular-weight β-amyloid oligomer levels are elevated in the CSF in AD (116). Yet in the cerebral tissue, the ratio of Aβ oligomer levels to plaque density distinguishes demented from non-demented patients (117).
Several membrane receptors can bind to Aβ oligomers. These receptors include the cellular prion protein (PrPC); the α7 nicotinic acetylcholine receptor (α7nAChR); Fcγ receptor II-b (FcγRIIb); the p75 neurotrophin receptor (p75NTR); the paired immunoglobulin-like receptor B (PirB); the PirB human orthologue receptor (LilrB2); the β-adrenergic receptors (β-ARs); and the Eph receptors (118), among others. PrPC is one of the binding partners for Aβ oligomers (119-125), and PrPC mediates impairment of synaptic plasticity by Aβ oligomers (124).
Besides β-amyloid species, several molecules are also components of SPs including metal ions, lipids, mucopolysaccharides, immunoglobulins, members of the complement system, molecules linked to lipid metabolism and lipid transport, blood coagulation/haemostasis factors, proteins linked to metabolism and molecular transport, neural, cell adhesion and extracellular matrix proteins, proteoglycans, and other cellular proteins (126, 127). The large amount of proteins in SPs is likely the consequence of co-aggregation and alteration of associated biochemical processes by which β-amyloid formation leads to neurodegeneration (127). Moreover, PrPC co-localizes with Aβ in SPs (128).
β-amyloid plaques are associated with variable alteration of neuronal processes, reactive astrocytes, and microglia. Altered synaptic protein deposition with a granular pattern is found in diffuse plaques (129). Neurotransmitter-containing and peptidergic dystrophic neurites precede those containing paired helical filaments within SPs (130-132). Altered neuronal structure, accumulation of abnormal molecules, and abnormal organelles and debris are characteristic of dystrophic neurites of mature SPs (133, 135). In addition to synaptic proteins, components of dense-core vesicles accumulate in dystrophic neurites of SPs (129, 135-138). Immunohistochemical studies have shown that dystrophic neurites of SPs contain 3Rtau and 4Rtau; several phospho-tau species; MAP2-P; phosphorylated neurofilaments light; medium and heavy chains; and active kinases p38, SAPK/JNK, GSK3β, and CK1-δ, in addition to markers of the ubiquitin-proteasome system (UPS) and autophagy (139). Mitochondria are altered in dystrophic neurites of SPs with variable vulnerability of the mitochondrial complexes of the respiratory chain (140, 141).
Dystrophic neurites are likely derived from axons arising from diverse neuronal populations, as revealed by specific neuronal markers (130-132, 142-146) therefore indicating that neuronal vulnerability is not restricted to a single cellular population.
Pyramidal cells in the vicinity of SPs show distorted dendrites and loss of dendritic spines (147-149).
2b. Tau
The microtubule-associated protein tau, encoded by MAPT, participates in microtubule stability, cellular polarity, and anterograde and retrograde axonal transport of organelles and vesicles. In addition to microtubules and actin, tau interacts with a large number of proteins and lipids in the cytoplasm, cell membranes, and synapses, and with DNA and proteins involved in DNA protection, among many other substrates (150, 151). The various functions of tau require interaction with multiple partners (151-157).
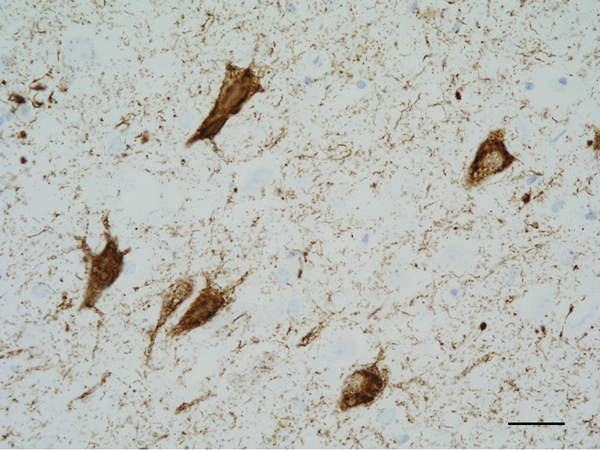
Figure 5: Neurofibrillary tangles in the CA1 region of the hippocampus. Paraffin section, AT8 immunohistochemistry, slight haematoxylin counterstaining, bar = 25μm.
The main constituent of NFTs dystrophic neurites of SPs and neuropil threads is abnormal tau (158-173) (Figures 5, 6, and 7). A combination of all six hyper-phosphorylated brain tau isoforms (3Rtau and 4Rtau expressed in brain), generated from alternative tau splicing, is characteristic of AD tau (163, 174, 175). The amount of 3Rtau is similar to 4Rtau in the human adult brain and in AD. However, possible variations in the ratio of 3Rtau/4Rtau among cell types in the human brain have not been adequately assessed. Abnormal tau in AD includes several species resulting from hyper-phosphorylation at different sites, acetylation, glycosylation, altered confor mation, truncation at glutamic acid 391 and at aspartic acid 421 (mediated by caspase 3), oligomerization, and β-sheet-rich fibril aggregation, among others (171-173, 176-196). The site of tau phosphorylation and other post-translational modifications in tau have commonalities and differences among tauopathies (197, 198). Tau inclusions in glial cells are not found in AD, unless accompanied by other tau co-morbidities including aging-related tau astrogliopathy (ARTAG) and argyrophilic grain disease (AGD) which are 4Rtau-only tauopathies.
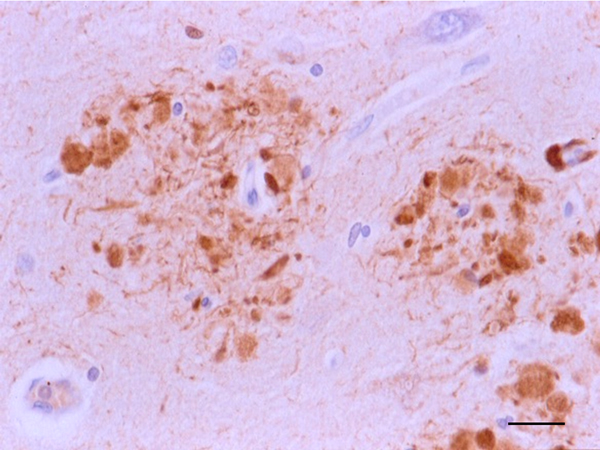
Figure 6: Dystrophic neurites of SPs in the entorhinal cortex containing hyper-phosphorylated tau. Paraffin section, AT8 immunohistochemistry, slight haematoxylin counterstaing, bar = 25μm
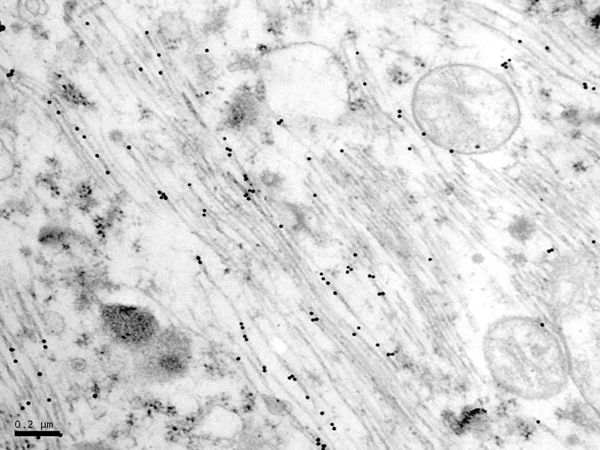
Figure 7: Immunoelectron microscopy showing phospho-tau deposits (black dots) in paired helical filaments. AT8 antibody, bar = 0.2μm.
Tau hyper-phosphorylation, the first step in NFT formation, is geared by the activation of specific kinases, and probably also by accompanying inhibition of phosphatases (171, 199). Several kinases are implicated in both the physiological and the pathological phosphorylation of tau, including glycogen synthase kinase 3β (GSK3β); cyclin-dependent kinase 5 (CDK5); protein kinase A (PKA); JUN N-terminal kinase (JNK); p38; and others (200). Co-localization of selected active kinases and tau deposits can be visualized in brain tissue (201-203). G-protein-coupled receptor (GPCR) kinases are also associated with NFTs and β-amyloid plaques in AD (204).
These alterations are cumulative but not homogeneous. More than one tau species may be present in a particular neuron. Furthermore, distinct defects may result depending on the type of accumulated tau, ranging from reversible dysfunction to irreversible disruption of the cytoskeleton, altered axonal transport, undermined cell signaling, synaptic dysfunction, and cell death. Tau-linked alterations can be the direct result of toxic species or the interactions of multiple partners (156). Soluble and insoluble tau oligomers, both phosphorylated and non-phosphorylated, may be involved in neurodegeneration (191, 205).
The association of tau with the plasma membrane is determined by its phosphorylation pattern. Tau associated with the plasma membrane can move to the cytosol upon tau hyper-phosphorlation (206, 207). The phosphorylation of tau also depends on phosphatidyl choline and phosphatidyl serine (208). Therefore, the composition of lipids at the membrane may modify the phosphorylation of tau and its capacity to shift its binding with actin and cytosolic proteins (209-211). Tau interactions with the membrane have several implications (155, 212). In addition to stabilizing membrane-cytosol interactions, tau is secreted associated to vesicles, or vesicle-free, key features in tau transmission (213-215).
Morphologically, abnormal neuronal tau deposits in AD are manifested as perinuclear tau deposits, granular cytoplasmic deposits, diffuse cytoplasmic deposits (all considered pre-tangle stages), neurofibrillary tangles (classical NFTs), ghost tangles (remains of NFTs in the neuropil), dystrophic neurites of SPs, and neuropil threads. The redistribution of abnormal tau from axons to the somatodendritic compartment of neurons and dendritic spines is a characteristic consequence of tau pathology in AD and other tauopathies. PHFs induce tau accumulation into aggresomes that gather misfolded proteins when the protein degradation system is overloaded (216).
The structure of tau filaments in the different tauopathies largely depends on tau composition (3Rtau and 4Rtau) and on post-translational modifications including conformation and truncation, as revealed by transmission electron microscopy, and more recently by optimized cryo-electronmicroscopy and mass spectrometry (193, 217-222). The different structure of tau aggregates in tauopathies indicates the formation of different tau strains which are specific to each tauopathy (223). The accumulation rate of tau aggregates is greater in females and younger β-amyloid-positive subjects (224). Increased expression of 19 genes in chromosome X is associated with tau burden and slower cognitive decline in women but not in men, suggesting that specific X chromosome factors could confer risk or resilience in aging and AD (225).
In addition to abnormal tau, NFTs contain numerous proteins. Total tau interacts with a good number of proteins in AD (226, 227). Laser-capture micro-dissection of NFTs and liquid chromatography-/tandem mass spectrometry (LC-MS/MS) analysis in sAD followed by affinity purification mass spectrometry revealed that seventy-five proteins present in NFTs interacted with PHF1-immunoreactive phosphorylated tau (228). NFTs also contain markers of the sequestosome/p62, ubiquitin, and mutant ubiquitin (229, 230).
Increased PrPC expression downregulates tau protein (231-234). Conversely, reduction or ablation of PrPC levels induces an increase in tau 3Rtau/4Rtau balance through downregulation of GSK3β activity, thus indicating that PrPC plays a role in tau exon 10 inclusion through the inhibitory capacity of GSK3β (235). Increased PrPC levels at early and middle stages of NFT pathology yields lower tau and hosphor-tau. In contrast, PrPC levels decrease at advanced stages of NFT pathology, which correlates with increased amounts of tau and hosphor-tau. Taken together, these observations suggest a protective role for PrPC in early stages of AD (236). These observations linking an interaction of prion protein and tau may have implications in certain familial prion diseases grouped under the term Gerstmann-Sträussler-Scheinker disease, in which abundant PRPRes-amyloid deposits are accompanied by extensive tau pathology (see section 3).
3. Familial AD (fAD; early-onset familial Alzheimer’s disease: EOFAD), and the β-amyloid cascade hypothesis
From the early nineties, mutations in the genes APP (β-amyloid precursor protein), PSEN1 (presenilin1), and PSEN2 (presenilin2), all of them involved in the production of β-amyloid, have been identified in several families with pre-senile dementia of Alzheimer’s type (early-onset familial Alzheimer disease: EOFAD, or fAD); increased APP dosage was also causative of AD and β-amyloid angiopathy (237-244). Recent genetic studies of the first Alzheimer’s case identified that the patient carried a mutation in PSEN1. These groundbreaking discoveries led to the “β-amyloid cascade hypothesis”, which supports the idea that the production of β-amyloid species is the primary factor triggering NFT formation and AD progression (245). The amyloid cascade hypothesis was further supported by the production of β-amyloid in transgenic mice bearing human mutations causative of AD. Yet mutations in these genes did not result in NFT formation in transgenic mice, although a few small hyper-phosphorylated tau deposits did appear in dystrophic neurites around β-amyloid plaques. At most, the joint production of SPs and tau deposits similar to NFTs in mice requires the cumulative expression of different mutated genes involved in human AD and tauopathies (246-248). However, in vitro and in vivo studies have shown the capacity of β-amyloid to phosphorylate tau and enhance tau aggregation, thus giving a boost to the β-amyloid cascade hypothesis (249). The β-amyloid cascade hypothesis fits with fAD linked to mutations of APP, PSEN1, and PSEN2.
Another genetic condition linked to increased risk of AD is Down syndrome. Middle-aged individuals (MA) with Down syndrome have neuropathological lesions of AD. Amyloid deposits may start as early as 12 years of age and they are universal by the age of 31. NFTs appear later in the entorhinal cortex, hippocampus, and neocortex (250-253).
The discovery of β-oligomers and cumulative evidence of their toxicity has led to modification of the “β-amyloid cascade” hypothesis, leading to the “amyloid-β oligomer hypothesis” (103-108). According to the new proposal, it is not the presence of fibrillar β-amyloid and deposition into definite aggregates, but rather soluble β-oligomers that are causative of cell damage and that trigger the process of neurodegeneration in AD (103-106, 254-257).
However, several points are still obscure. The level of insoluble Aβ rises with age and is further increased in AD whereas the total level of Aβ40 in both soluble and insoluble fractions and the level of Aβ42 in the soluble fraction decline with age before about 50 years. Differential production or retention of Aβ40 and Aβ42 likely contributes to the influence of age on the risk of sporadic AD, but the levels of soluble Aβ concentrations, higher in young adults than in older individuals and in subjects with AD, do not match the proposed toxic role of oligomers in AD (258, 259).
Tau deposits, other than those located in dystrophic neurites of SPs, are largely independent of β-amyloid. Other factors, including apolipoprotein E (ApoE), the endocytic system, cholesterol metabolism, and microglial activation, are regulators of tau pathology (260). Neurons derived from induced pluripotent stem cell (iPSC) lines from sAD and fAD linked to PSEN1 mutations show increased phosphorylation of tau at different sites, increased levels of active GSK3β, and a significant upregulation of APP synthesis and APP carboxy-terminal fragment cleavage. However, significantly increased Aβ1-42/Aβ1-40 ratios are observed in fAD but not in sAD (261).
Other amyloids are the main constituents, in combination with NFTs, of different genetic neurodegenerative diseases causing dementia. Familial British dementia (FBD) and familial Danish dementia (FDD) are linked to specific mutations in the BRI2 gene; the cleavage of Integral membrane protein 2B (BRI2) produces ABri and ADan amyloidogenic peptides, respectively. Amyloid plaques and amyloid angiopathy, and NFTs with a tau composition identical to AD tau, are found in both diseases (262, 263). Gerstmann-Sträussler-Scheinker disease (GSS) is linked to mutations in the prion protein gene (PRNP) that cause a prionopathy. Depending on the muta-tion, GSS is manifested pathologically by a combination of abundant prion-immunoreactive plaques surrounded by dystrophic neurites, together with numerous NFTs indistinguishable from AD-NFTs (264-266). Interestingly, APP, BRI2, and prion are proteins located at the cell membrane, and they interact with each other in normal conditions. The non-fibrillar, soluble BRI2-derived amyloids are also toxic, and probably play a central role in the pathogenesis of BRI2-linked dementias (267). The common structure of soluble amyloid oligomers suggests a common mechanism of pathogenesis (109, 113). Despite the differing genetic nature of these disorders, plaques and NFTs do not appear until middle age. Understanding of the mechanisms that control the metabolic pathways, that delay the beginning of the molecular and clinical manifestations of the disease for years, is a major challenge in neurodegenerative diseases linked to mutations in specific genes.
In contrast to mutations linked to β-amyloid production, mutations in MAPT are causative of familial tauopathy and are never associated with β-amyloid or other amyloid deposits (170, 171).
4. Sporadic AD (sAD; Late-onset Alzheimer disease: LOAD)
Most cases of AD (more than 95%) are sporadic (sAD) and occur in older individuals (late-onset Alzheimer’s disease: LOAD). sAD has an insidious onset and a progressive course leading to death about 10-15 years after the first clinical symptoms of dementia. Aging is the main contributory factor. sAD is favoured by individual or combined low penetrating genetic factors, mainly allele ε4 of ApoE (268-271). Genome-wide association studies (GWAS) have identified other risk genes of sAD: LDL receptor related protein 1 (LRP1); low density lipoprotein protein receptor 1 (LDLR); interleukin 1a; clusterin (CLU); phosphatidylinositol binding clathrin assembly protein (PICALM); complement component (3b/4b) receptor 1 (CR1); bridging integrator 1 (BIN1), involved in synaptic vesicles and endocytosis; triggering receptor expressed on myeloid cells 2 (TREM2); sortilin-related receptor 1 (SORL1), involved in endocytosis and sorting; ADAM metallopeptidase domain 10 (ADAM10), involved in the cleavage of several proteins; ATP binding cassette subfamily A member 7 (ABCA7); Spi-1 proto-oncogene (SPI1); paired immunoglobin like type 2 receptor alpha (PILRA); membrane-spanning 4-domains subfamily A (MSA4), linked to inflammation; CD2-associated protein (CD2AP) that regulates actin cytoskeleton; and ephrin receptor A1 (EPHA1), among others (272-288).
The regional and areal distribution of NFTs and SPs in the cerebral cortex is not homogeneous. In the hippocampal complex, NFTs predominate in the CA1 region and subiculum, the CA2, CA3 and hilus are less affected, and the dentate gyrus is spared in pure sAD cases. In the entorhinal cortex, NFTs are more abundant in layers II and V, whereas in the neocortex, NFTs predominate in layers III and V, with marked regional variations (the primary motor and sensory cortices have fewer NFTs than the association areas). NFTs are found more abundant in the temporal cortex, followed by the frontal and parietal cortex, and the occipital cortex. In subcortical regions, NFTs are localized in the basal nucleus of Meynert and nuclei of the basal forebrain, amygdala, hypothalamic nuclei, relay neurons within intralaminar and limbic thalamic nuclei, ventral tegmental area, raphe nuclei, locus ceruleus, and olfactory bulb. The cerebellar cortex is spared of NFT pathology. Cortical neurons with NFTs are mainly subpopulations of large pyramidal glutamatergic neurons (289-291). This is consistent with the observation that neurons with high content of neurofilaments are more susceptible to NFT formation (290-295).
GABAergic neurons are more resistant to NFT pathology, although the density of GABAergic neurons decreases and GABA-uptake is impaired in sAD (296-300) (see section 6 for details). Somatostain, which is expressed in a subpopulation of inhibitory neurons, and somatostatin receptors are also reduced in sAD (296, 301, 302).
Calcium-binding proteins parvalbumin (PV), calbindin D28K (CB) and caretinin (CR) are expressed in subpopulations of GABAergic neurons (303-305). PV-positive neuron numbers in the temporal, visual, and prefrontal cortex are preserved in AD (306-309), but PV-immunoreactive neurons are decreased in the entorhinal cortex and hippocampus in sAD (310-316). CB-immunoreactive neurons in the hippocampus, entorhinal cortex, and cortical layers V and VI are vulnerable, whereas CB-positive neurons in the occipital cortex and upper layers of the frontal cortex are resistant (309, 314, 314, 317). CR-positive neurons are not affected in the prefrontal, temporal, and visual cortices (309, 319, 320), but their number is reduced in the hippocampus and entorhinal cortex (314, 321). PV and somatostin, together with neuropeptide Y, cholecystokinin and substance P, are found in dystrophic neurites of SPs, thus evidencing the involvement of inhibitory and peptidergic neurons in SPs (130, 144).
Neuron loss is negligible in cognitively normal subjects, but the number of neurons decreases in the hippocampus and entorhinal cortex with NFT progression (322-324). The rate of this process is extremely variable among individuals.
A major achievement in improving our understanding of the progression of sAD pathology was the staging of NFTs and SPs in the post-mortem brain of large cohorts of non-demented and demented individuals covering a natural human population. In the telencephalon, the first NFTs appear in the entorhinal and transentorhinal cortex (stages I-II), followed by the hippocampus, temporal cortex, and other nuclei of limbic system (stages III-IV), and then continue on to most areas of the neocortex (stages V-VI). The spreading of NFTs is accompanied by a dramatic increase in the number of neurons with NFT pathology across stage progression (41, 325-327). About 85% of individuals aged 65 have NFT pathology, at least restricted to stages I-III (41, 325, 328, 329). All of them, excluding those having concomitant pathologies, are considered “cognitively normal for age” (330). Some individuals at stage IV-V suffer from moderate cognitive impairment; only about 5% have dementia. However, dementia of AD type accounts for about 25%-30% of the population at the age of 85 years, all of them categorized as NFT stages V-VI (331). Regional- and stage-dependent neuropathological alterations in sAD are accompanied by specified patterns of altered gene expression, that extend beyond the genes implicated in tau and β-amyloid pathology (332).
The Braak staging scheme does not rule out the occurrence of exceptions that do not fulfil the strict neuropathological criteria. These untypical cases are considered AD subtypes: hippocampal sparing, limbic-predominant, and minimal atrophy sAD subtypes might account for about 25% of cases (333). In addition, several clinical sAD variants including non-amnestic, corticobasal syndromal, primary progressive aphasia, posterior cortical atrophy, behavioral/dysexecutive, and mild dementia variants have been categorized (334).
The olfactory bulb and tract, and several nuclei of the brain stem including the raphe nuclei and the locus coeruleus, are affected by NFT formation at the first stages of NFT pathology. The involvement of the olfactory bulb and tracts may contribute to the altered olfaction arising in sAD. Damage to selected nuclei of the brainstem, which are the origin of major serotoninergic and noradrenergic innervation of the entire brain, underlies a large series of clinical symptoms including impaired arousal, loss of attention and memory, impaired decision making, apathy, depression, anxiety, and altered reward processing, among others (335-340). Considered together, NFT generation and neuron loss largely depend on the specific cell and regional vulnerability of specific neuronal populations. Moreover, the simultaneous presence of NFT lesions in separate brain regions indicates that there is no single origin of NFT pathology that spreads through the brain, but rather various and cumulative original sources of tau pathology in the aging brain. Additionally, the rates of NFT progression, although variable from one individual to another, appear slowly at early NFT stages and progress rapidly at advanced stages of the disease (41).
The distribution of SPs differs from NFTs in the cerebral cortex in patients with sAD (341). Assessing the same series of cases for the study of NFT progression evidenced that the localization and distribution of SPs largely differ from NFT staging. The majority of individuals at NFT stages I-II and almost half of those at stage III do not have SPs or β-amyloid deposits (41, 342). Stages 0, A, B, C of Braak define the progression of SPs through the neocortex. The phases of Thal represent, from phase 1 to 6, the progressive and cumulative appearance of SPs from the neocortex, allocortex, diencephalic nuclei, striatum, and cholinergic nuclei of the basal forebrain, the brainstem, and the cerebellum (343).
The early appearance of tau pathology compared with the later appearance of β-amyloid plaques in a series of 2366 cases from children to centenarians has been recently revisited (344). Based on the results of these observations and many previous studies, the paper hypothesizes that tau pathology is an initiating factor in sAD (344).
Indeed, the lack of temporal and regional concordance between NFTs and SPs in sAD is intuitively barely consistent with the β-amyloid cascade hypothesis, unless non-identified soluble or other species of β-amyloid interact with neurons, thus triggering NFT pathology (345).
These comments do not mean that there is no interaction between the two proteins. Tauopathy fuelled by β-amyloid in a synergetic mechanism is well documented in AD (346-348).
The arguments between supporters of β-amyloid and of tau as the primal origin of sAD have consumed a great deal of effort, time, and financial investment (349). There is no doubt about the new acquisition of knowledge generated regarding sAD pathogenesis, but exclusive hypotheses have not produced the anticipated unequivocal results.
Cognitive impairment and dementia correlate with tau deposition and NFT pathology rather than with β-amyloid deposits and SPs (331, 350-358). Neuron loss occurs largely in parallel with tau pathology rather than with SPs in most regions (359). However, neuron degeneration and neuron loss are not restricted to neurons with NFTs (see section 10).
Recently, a classification of AD has been proposed: AD autosomal dominant (fAD), ApoEε4 sAD, and non-ApoEε4 sAD (360). This categorization is not new, but rather recovers and further emphasizes the well-known relative importance of β-amyloid deposition in the different AD categories depending on genetic factors involved in the production of β-amyloid.
5. NFTs and SPs in non-human brain aging
β-amyloid plaques and β-amyloid angiopathy may be found in old-aged animals in some species including non-human primates, monkeys, dolphins and other cetaceans, dogs, cats, bears, and pinniped species, among others; deposits are usually diffuse whereas core plaques surrounded by tau-containing dystrophic neurites are exceptional (361-372).
Phosphorylated-tau deposits in neurons are rarely encountered in most mammals, and they usually have the characteristics of pre-tangles rather than NFTs, as in a few vulnerable aged mouse lemurs (362). Intracytoplasmic tau inclusions in neurons, astrocytes, and oligodendrocytes may occur in aged baboons (373, 374), aged gorillas (369), and chimpanzees (371). Tau accumulation in the brain of old sea lions, seals, and walruses forms argyrophilic fibrillar 3Rtau and 4Rtau aggregates in the neuronal somata and neurites, and olny few tau aggregates are found in oligodendrocytes and microglia (372). Importantly, these changes are linked to aging, but they are not the only expression of brain aging (375, 376).
Hyperphosphorylated tau accumulation in neurons, intraneuronal β-amyloid deposits, and diffuse amyloid plaques may occur in the brain of aged domestic cats (377, 378). The characteristics and distribution of tau lesions in a few cats are reminiscent of sAD including the deposition of 4Rtau and 3Rtau (379). A unique 4Rtauopathy without β-amyloid deposits mainly involving neurons of the neocortex but not the hippocampus, accompanied by widespread coiled bodies in the cerebral white matter, has been reported in aged domestic cats (380).
These observations show that β-amyloid deposition and tau pathology may occur with high species variability, in aged mammals, and, particularly, in non-human primates and pinnipeds. However, we do not have evidence at present on whether these species show changes in the same way as human beings. In aged cynomolgus monkeys, β-amyloid plaques combine with 4Rtau deposits in pre-tangle neurons and coiled bodies in glial cells with a regional pattern reminiscent of progressive supranuclear palsy (370). Therefore, the old hypothesis suggesting that sAD is a phylogenetic disease (381) has a relative relevance unless applied to the search for mechanisms modulating similarities and differences between non-humans and humans regarding the tremendous prevalence, widespread localization, particular regional distribution, and composition and structure of tau deposits in humans in comparison with other species.
6. Synapses
Synaptic alterations were described in the 1960s in the seminal electron microscopic studies of AD (382). These findings were followed by the observation of decreased numbers of dendritic spines on cortical neurons assessed with the Golgi method in post-mortem and biopsy samples at a time when cerebral biopsies were still considered appropriate tools for diagnosis of dementia (383-389). Synaptic loss is the major morphological correlate of cognitive impairment (390). For this reason, AD is considered as the consequence of a synaptic failure (391). Subsequent studies have refined synaptic alterations using different methods (392; 393), including the use of intraneuronal dyes in post-mortem tissues (394, 395).
The Golgi method also provided evidence of dendritic degeneration and dendritic sprouting and re-growth in several brain regions in AD (396-401). Dendritic sprouting is reinforced by the presence of growth-associated protein 43 (GAP-43), a marker of neuritic growth and sprouting around SPs (143, 402). Aberrant sprouting seems to be triggered by pre-amyloid species and neurotrophic factors (401). Aberrant sprouting involves neurites, dendrites, and synapses, and it affects distinct connections in AD (401). Cycles of aberrant synaptic sprouting and neurodegeneration are common in AD (403).
Spine loss occurs mainly in clusters linked to tau pathology (404, 405). Immunohistochemistry also reveals altered expression of synaptic markers not only around SPs but also in diffuse plaques, suggesting a close relationship between synapses and β-amyloid deposition (129, 406, 407). Abnormal pre- and post-synaptic tau and tau oligomers damage the synapses and produce altered synaptic function (150, 408, 409).
Double-labeling of neurons also shows a direct relationship between tau deposition and loss of dendritic spines on cortical pyramidal neurons in AD (404). Abnormal tau and β-amyloid oligomers act synergistically to disrupt synaptic function (409). However, synaptic loss also appears not to be dependent on fibrillar β-amyloid in a murine model of β-amyloid deposition (149). Abnormal neuronal expression of APP and cytoskeletal proteins in early stages of the disease might be involved in the mechanisms of synaptic pathology in AD (410).
Synaptic proteins are important components associated with β-amyloid in SPs (127). It has recently been postulated that altered synapses are the origin of amyloid plaques in AD (411; see also section 2a). Both β-amyloid and abnormal tau are accumulated at the synapses (412-421). Recent neuroimaging studies further support the association of tau pathology, synaptic loss, and altered synaptic function (422). It has been proposed that synaptic tau pathology is an early event, and synaptic tau seeding precedes tau pathology in sAD (423). Other factors are also important such as cytoskeletal actin dysregulation (424), and oxidative stress lipid and protein damage (425). No less significant is the association between cell-cycle dysfunction and failure of synaptic plasticity in AD (426).
Synaptic alterations include abnormalities in the synaptic and postsynaptic delivery of neurotransmitters and neuromodulators, and the selective vulnerability and responses of their receptors (see section 7).
Finally, lipid and protein alterations at the cell membrane, and altered cytoskeletal proteins, may affect synaptic integrity and function (see sections 22c and 22h).
In addition, synapses are organelles with high energy consumption, and therefore they are vulnerable to deficits in energy production linked to mitochondrial failure (427) (see section 22d).
7. Neurotransmitters, neuromodulators, and related receptors
An early relevant biochemical observation was the discovery of the involvement of the Meynert nucleus in AD, the correlation of this involvement with the number of plaques and cognitive impairment, and the accompanying impairment of cholinergic innervation in the cerebral cortex (428-430). The “cholinergic hypothesis” stated that AD was a disorder of cholinergic innervations (430, 431). The enthusiasm for the cholinergic theory was supported by the prior discovery of dopamine deficiency in the substantia nigra pars compacta in Parkinson’s disease, and the success of L-dopa treatment for this disorder which is still in use 50 years later (432). Cholinergic drugs were used although their benefits were clearly lower than initially expected.
Later, the glutamatergic theory stated that excitotoxicity resulting from excessive synaptic or extrasynaptic activation of N-methyl-D-aspartate (NMDA) subtype of ionotropic L-glutamate receptors might enhance vulnerability of neurons in AD (433, 434). The role of glutamate in the pathogenesis of AD was driven, in part, by the discovery of altered glutamate transport and increased excitotoxicity in amyotrophic lateral sclerosis (435), and the interest at that time in excitatory amino acid neurotoxicity in the pathogenesis of neurodegenerative diseases (436). Glutamate overload increases mitochondrial Ca2+ influx and oxidative stress and leads to mitochondrial dysfunction (437). However, NMDA receptor blockers may also have undesirable effects due to their double effects on cell death as well as cell survival and plasticity (438). Now, neuroprotective therapies aim to both enhance the effect of synaptic activity and disrupt extrasynaptic NMDAR-dependent death signaling (438).
Cholinergic and glutamatergic neurotransmitter alterations play a significant role in the pathogenesis of brain aging and sAD (439-442). Other neurotransmitters and receptors are involved as well.
The important point is that alterations are not homogeneous; they depend on the type of neurotransmitter, the cells of origin, and the kind of receptor; not all receptors of a given neurotransmitter are equally vulnerable to aging and sAD.
In addition to the neurotransmitters, neuromodulators, and receptors discussed below, general aspects of GPCR, amylin receptors, netrin receptors, and dopamine receptors in sAD are detailed in other reviews (443, 444). Endorphins, enkephalins, dynorphins, and endomorphins are endogenous opioid peptides that bind to opioid receptors. β-endorphin has opioid activity through μ-receptors, but α-endorphin and γ-endorphin lack affinity for opiate receptors. Endorphins interact with the γ- aminobutyric acid (GABA), which in turn modulates the release of dopamine. The expression of endogenous opioids and receptors is altered in human brain aging and sAD (445-448).
Many olfactory and taste receptors and molecules involved downstream are expressed in the human brain (449). Their ligands and functions remain unknown, although both olfactory and taste receptors might contribute to intercellular and intracellular cell signaling. The expression of some olfactory receptors is altered in sAD and other neurodegenerative diseases (450).
7a. Acetylcholine (Ach) and acetylcholine receptors (AChR)
ACh is synthesized in neurons by choline acetyl transferase (ChAT). ACh acts upon nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs). ACh is degraded by acetylcholinesterase (AChE). Nicotinic receptors are ionotropic ligand-gated receptors, and muscarinic receptors are GPCR (451). The nAChRs are arranged into homomeric or heteromeric subunits consisting of a diverse set of complex subtypes including α1-7, α9-10, β14, γ, δ, and ε. Allosteric modulation of nAChRs increases pre-synaptic ACh levels and enhances the cholinergic nicotinic neurotransmission. α7 and α4β2 nAChR mediate the presynaptic release of ACh. nAChRs are also expressed in astrocytes and microglia (452, 453).
Through its receptors TRkA and p75NTR, the nerve growth factor (NGF) plays an essential role in the survival and maintenance of cholinergic neurons in the basal forebrain (454-457).
Loss of basal forebrain cholinergic neurons occurs at early stages of NFT pathology (428, 458, 459). Pretangle pathology within cholinergic nucleus basalis neurons coincides with local neurotrophic and neurotransmitter receptor gene dysregulation (460). Loss of cholinergic neurons in the basal forebrain leads to reduced ACh levels and ChAT upregulation (461). In addition, nAChRs, particularly α7nAChRs, are altered in sAD (462-465). Importantly, nicotine and nAChRs also participate in the regulation of Aβ. On the one hand, nicotine inhibits the formation of Aβ1-42 fibrils and disrupts preformed Aβ fibrils (466). On the other hand, Aβ1-42 binds to α7nAChR and inhibits the release of ACh (467). Finally, α7 nAChRs mediate Aβ-induced neurotoxicity (468, 469). and Aβ-induced tau phosphorylation (470).
α7nAChRs also participate in microglial activation (471). Astrocytic and microglial nAChRs modulate Aβ phagocytosis and degradation, Aβ-related oxidative stress, and neurotoxicity (472).
The metabotropic mAChRs are classified into five M1-M5 subtypes (473). M1, M3, and M5 receptors interact with the Gq/11 protein, stimulate phospholipase C (PLC), phosphatidylinositol trisphosphate (PI3P), and activate protein kinase C (PKC). M2 and M4 receptors interact with Go/i proteins, inhibit adenylyl cyclase (AC) and protein kinase A (PKA), and decrease cAMP levels (474-476). There are no changes in the number of mAChRs in sAD; however, the interaction of Gq/11 protein is altered in sAD compared with controls (477). M1 muscarinic agonists reduce β-amyloid and tau pathology, whereas M1 muscarinic antagonists or deletion of M1 subtype augment β-amyloid and tau pathology in in vitro and in vivo murine models of AD (478-480).
SPs have low levels of AChE, but the activity of AChE increases around β-amyloid plaques (481, 482). AChE inhibitors such as donepezil, galantamine, tacrine, and rivastigmine are administered at the initial stages of sAD.
7b. Glutamate and glutamate receptors (GluRs)
Glutamate is released from synaptic terminals and acts on post-synaptic ionotropic glutamate receptors (iGluRs). This mechanism mediates fast excitatory synaptic transmission. Glutamate can also act on metabotropic glutamate receptors (mGluRs). The mechanism modulates various effects by coupling to G proteins with subsequent recruitment of second messenger systems. There are three families of iGluRs: NMDA (N-methyl-D-aspartate receptor), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), and KA (kainate) receptors: NMDAR, AMPAR, and KAR, respectively. NMDARs are composed of different subunits encoded by GluN1 (NR1), GluN2A (NR2A), GluN2B (NR2B), GluN2C (NR2C), GluN2D (NR2D), GluN3A (NR3A), and GluN3B (NR3B). AMPARs are composed of combinations of GluA1 (GluR1), GluA2 (GluR2), GluA3 (GluR3) and GluA4 (GluR4); and KARs by GluK1 (GluR5), GluK2 (GluR6), GluK3 (GluR7), GluK4 (KA-1) and GluK5 (KA-2). In NMDARs, the binding of glutamate and glycine is necessary to activate glutamate-gated ion channels. The removal of magnesium ions (Mg2+) block permits the entry of calcium ions (Ca2+) and synaptic signaling (483, 484).
Both iGluRs and mGluRs are also localized pre-synaptically acting as auto-receptors and hetero-receptors. This localization facilitates neurotransmission in the short term and depresses neurotransmission in the long term (485). GluRs are localized non-synaptically and are also expressed by astrocytes and oligodendrocytes (486).
Glutamate binding to NMDARs, AMPARs, and KARs is reduced in the aging brain and sAD mainly in the cerebral cortex and hippocampus. These changes are receptor-, region-, and layer-dependent, thus indicating variable vulnerability. Altered expression is likely dependent on several factors and not necessarily correlated with local NFTs and SPs, although loss of neurons and reduced neuronal connectivity may account, in part, for the decreased receptor expression (440). The localization of the receptors also plays a cardinal role. NMDARs containing GluN2A subunits are located at synaptic sites and are implicated in the protective pathways. In contrast, GluN2B subunits are located mainly at extra-synaptic sites, and they increase neuronal vulnerability. β-amyloid activates GluN2B-containing NMDARs (487-489). In addition, NMDARs are necessary for synaptic targeting of Aβ oligomers (490) and neuronal Aβ production (491). NMDAR alterations are implicated in synaptic dysfunction in sAD (492, 493). NMDARs also participate in redox-mediated synaptic function impairment in brain aging and sAD (494).
Memantine, an NMDAR antagonist, is currently used at the middle clinical stages of AD to reduce the hyperactivity of glutamate, resulting in transient and limited success.
AMPARs are consistently endocytosed. The increase in the rate of AMPA endocytosis induces long-term depression and synaptic degeneration (495). Soluble Aβ oligomers are involved in synaptic damage via the subunit GluA3 AMPAR (496). Aβ also induces AMPAR ubiquitination and degradation (497).
Glutamate abnormalities in aging and sAD are not restricted to alterations in glutamate production and its effects on synaptic receptors in neurons and glial cells. Furthermore, glutamate effects in normal and pathological conditions also depend on glutamate transport by specific neuronal and glial transporters (498).
Excitatory amino acid transporters (EAATs) re-uptake glutamate from the synaptic cleft and extra-synaptic sites, and transfer glutamate to glial cells and neurons. Vesicular glutamate transporters (VGLUTs) move glutamate from the cell cytoplasm into synaptic vesicles. EAATs can also transport L-aspartate and D-aspartate. EAAT1 and EAAT2 (SLC1A3 and SLC1A2, respectively) are localized in astrocytes whereas EEAT3 (SLC1A1), EAAT4 (SLC1A6) and VGLUTs 1/2/3 (SLC17A7, SLC17A6, SLC17A8, respectively) are found in neurons. Loss of EAAT2 occurs in many neurodegenerative diseases, including sAD (see section 12a). Abnormal expression of VGLUTs and EAATs may contribute to neuronal excitotoxicity and neuron demise in sAD (499-501).
mGluRs are G-coupled proteins that act upon different effector systems, including PLC and AC. mGluRs are classified into three groups based on their pharmacological profiles, molecular properties, and transduction mechanisms. Group I receptors (mGluR1, mGluR5) are coupled to PLC activation through Gq/11 proteins, whereas groups II and III are coupled to AC inhibition through Gi/o proteins. mGluR1/5 are primarily excitatory, and mGluR2/3 and mGluR4/5/6,7/8 are inhibitory (502-505). mGluRs, determined by radioligand binding assays, and expression levels of mGluR1, detected by western blotting, are significantly decreased in the frontal cortex in sAD. This decrease is already observed at NFT stages I-II and III-IV not involving the frontal cortex, and further decrease with the appearance of NFTs in the frontal cortex with disease progression. The expression levels of phospholipase Cβ1 (PLCβ1) isoform, which is the effector of group I mGluRs, is decreased in parallel. PLCβ1 decrease, in turn, is associated with reduced GTP- and l-glutamate-stimulated PLC activity in sAD. These results show that group I mGluRs/PLC signaling is downregulated and desensitized in the frontal cortex at the first stages of NFT pathology, and that these modifications worsen with the progression of sAD (506).
7c. γ-aminobutyric acid (GABA) and GABA receptors
The inhibitory neurotransmitter GABA is generated by α-decarboxylation from L-glutamate in neurons by the action of glutamic acid decarboxylase (GAD). GABA is then incorporated into the synaptic vesicles by vesicular GABA transporter (VGAT). After release from synaptic vesicles, GABA binds to ionotropic GABAA and metabotropic GABAB receptors. GABAA receptor activation opens chloride ion channels; GABAB acts through G proteins, reduces calcium ion channels, and inhibits AC and intracellular production of cAMP (507-510). GABAA receptors are composed of combinations of five different subunits, α1-α6, β1-β3, γ1-γ3, δ, ε, θ, π, and ρ1-ρ2. The most frequent pentamers are 2α:2β:1γ (507). GABAB receptors are heterodimers composed of R1 and R2 subunits (508). GABA at the synapses is picked up by astrocytes that catalyze it to glutamine, which is then transported into neurons and converted to glutamate (511, 512). The involvement of astrocytes in GABA metabolism has suggested a potential role of astrocytes in GABA gliotransmission (513).
Several studies have shown inconsistent results regarding total GABA levels in sAD but there is a trend toward stressing abnormal GABAergic function (514-516). Reduced GABA, glutamate, and glutamine levels are observed in individuals with MCI and AD as revealed by magnetic resonance spectroscopy (517-520).
Regarding GABA receptors, electrophysiological studies reveal a reduction in GABA currents in the temporal cortex in sAD. This is associated with mRNA downregulation of α1 and γ2 subunits and upregulation of α2, β1, and γ1 transcripts (521). α1 and α5 subunit protein-immunoreactive levels are decreased in the CA1 region of the hippocampus (521-524), whereas α3, β1, β2, β3, and γ2 subunits are unaffected. In contrast, α1 subunits are increased in the CA3 region, granule cell layer, and hilus of the dentate gyrus in sAD (523-527). β3 subunit expression is decreased in the stratum oriens, radiatum of CA2 and CA3, and stratum moleculare (527), whereas γ1/3 subunits are upregulated in the hippocampus in sAD (525, 527).
Aβ induces the downregulation of GABAA receptors, inhibitory dysfunction, and sprouting of GABAergic axons (528-532).
7d. Serotonin and 5-hydroxytryptamine (5-HT) receptors
5-Hydroxytryptamine (5-HT) derives from the amino acid tryptophan via the intermediate 5-hydroxytryptophan and decarboxylation to form serotonin. In the brain, 5-HT is mainly produced in the raphe nuclei of the brain stem that constitutes part of the reticular formation. Ascending serotoninergic fibres innervate the whole telencephalon. Descending projections innervate the cerebellum and the spinal cord (443). Serotonin is stored at the synaptic vesicles and released into the synapse, where it binds to post-synaptic and auto-pre-synaptic receptors. Serotonin is then re-uptaken via serotonin transporters and reused or degraded by monoamine oxidase. 5-HT receptors are categorized into metabotropic and ionotropic receptors. Metabotropic GPCR are 5-HT1, 5-HT2, 5-HT4, 5-HT5, 5-HT6, and 5-HT7. The only ionotropic receptor is 5-HT3, permeable to sodium, potassium, and calcium ions. 5-HT1 and 5-HT5 receptors bind to Gαi/o proteins, inhibit AC, and decrease cAMP levels. 5-HT2 receptors bind with Gαq/11, activate PLC, generate PI3P, and activate PKC (533-541).
5-HT and receptors interact with the cholinergic, glutamatergic, noradrenergic, GABAergic, endocannabinoid, and glial cell systems (542-545). Orexins regulate serotonin neurons in the raphe nucleus (546).
The serotoninergic system is altered in aging and sAD (443, 547-549). The main alteration of the serotoninergic system in brain aging and sAD results from neuronal damage and NFT formation in the raphe nuclei in the independent origin of NFT pathology at early stages of AD-related pathology. Damage to the serotoninergic system contributes to mood changes and depression which are characteristic non-cognitive clinical manifestations of brain aging and sAD (443, 550-553).
Serotonin is linked to decreased β-amyloid production and modulation of soluble β-amyloid precursor protein (sAPPβ) (554-557). In addition, 5-HT4 receptors inhibit the secretion of β-amyloid peptides (558-560).
Due to the multiple facets of serotonin, serotonin receptors, and their interaction with other neurotransmitters, agonists, antagonists of the different 5-HT receptors, and principally selective serotonin re-uptake inhibitors are useful pharmacological agents to improve cognition and reduce depression in aging and sAD (444, 552, 561-568).
7e. Noradrenergic system
Norepinephrine or noradrenaline is synthesized from dopamine by the enzyme dopamine β-hydroxylase (DBH). Norepinephrine is metabolized by mono-amino oxidase (MAO) and catechol-O-methyltransferase (COMT). Norepinephrine is transported from the cytosol to the synaptic vesicles by the vesicular monoamino transporter (VMAT) (569, 570). Norepinephrine can bind both to metabotropic pre- and post-synaptic α1, α2, β1, β2, and β3 receptors. α1 are Gq-coupled and activate PLC; α2 are coupled to Gi/G0 proteins and inhibit AC; β1, β2, and β3 are coupled to Gs proteins and activate AC (571-574).
The locus coeruleus, which contains about 15,000 neurons in primates, is the principal source of brain noradrenaline. Noradrenergic terminals innervate the hippocampus, amygdala, cerebral neocortex, and hypothalamus (575-577). Post-synaptic α1 receptors are excitatory, whereas perisomatic and pre-synaptic α2 receptors are inhibitory (578). Adrenergic receptors are widely distributed in the brain (579, 580). Neurons of the locus coeruleus excite the cerebral cortex principally through α1 receptor signaling (581, 582). The locus coeruleus-noradrenergic system has a major role in arousal, attention, and stress responses. In the brain, norepinephrine may also contribute to long-term synaptic plasticity, pain modulation, motor control, and energy homeostasis (583). Noradrenergic terminals are also in contact with glial cells and blood vessels (584-586). Due to these connections, noradrenergic innervations also modulate inflammation and cerebral blood flow (583, 587-589).
In addition, the noradrenergic system interacts with the cholinergic and GABAergic systems (576, 590-592). Moreover, the locus coeruleus and the raphe nuclei are interconnected (593, 594). Finally, orexin/hypocretin, histamine and noradrenaline converge in the dorsal raphe nucleus (593).
The locus coeruleus is damaged at early stages of AD-related pathology (443, 595-598) (see also section 4). At these first stages, NFTs accompany neuron loss, but SPs only appear in some cases at advanced Thal phases of β-amyloid deposition. In contrast to the massive loss of noradrenergic neurons, calbindin-immunoreactive neurons are preserved in the locus coeruleus even at advanced stages of NFT pathology (599). Early neuronal alterations in the locus coeruleus are accompanied by abnormalities in the ascending noradrenergic system (600-602). Increased α2A adrenergic receptor protein occurs in the amygdala and hippocampus in parallel with early NFT pathology in the locus coeruleus (338). In contrast, reduced DBH activity is found in the post-mortem hippocampus and neocortex, probably as a compensatory mechanism to noradrenaline lessening (603, 604). DBH levels are also reduced in plasma at early stages of AD (605). Moreover, connectivity between norepinephrine and dopamine brainstem centers is disrupted in sAD (606).
The orexin system is compromised in sAD, thus contributing, in combination with the noradrenergic and serotonin decay, to altered sleep in sAD (607-611).
7f. Adenosine receptors
Adenosine is transported across the plasma membrane based on: a) its concentration gradients, and b) active Na+-dependent transporters that carry adenosine against its concentration gradient.
Adenosine receptors are purinergic GPCR classified into A1, A2A, A2B, and A3 receptors. A1 and A3 receptors inhibit AC through Gi/o proteins, while A2A and A2B receptors stimulate AC through Gs proteins (612, 613). Adenosine receptors are present in subpopulations of neurons, astrocytes, oligodendrocytes, and microglia (614-616). Adenosine receptors modulate the release of glutamate, GABA, acetylcholine, noradrenaline, and serotonin (616-623).
Early autoradiographic studies showed decreased A1 expression in the hippocampus at advanced stages of sAD (624-627). However, A1 receptors accumulate in neurons with NFTs in sAD (628). More recent studies at first stages of NFT pathology have shown upregulation of adenosine receptors and sensitization of their specific signaling pathways preceding NFTs and SPs in the frontal cortex (629).
7g. Endocannabinoids and cannabinoid receptors (CBRs)
CBRs are classified as type 1 (CB1R) and type 2 (CB2R) (630). Anandamide (N-arachidonoyl ethanolamine, AEA) and 2-arachidonoyl glycerol (2-AG) are the main endogenous ligands of CBRs (631-634). Both endocannabinoids derive from arachidonic acid (AA). They are synthesized and metabolized by different pathways and induce specific biological functions. In the human brain, CB1Rs are mainly expressed in the limbic system. CB1Rs localize in the pre-synapses modulating glutamate and GABA neurotransmission (635-641). CB2Rs are expressed in microglia, and participate in inflammation and phagocytosis (642, 643). Low levels of CB2R expression have also been identified in some neurons (644-646).
Cannabinoid compounds may also bind to other receptors, such as GPR55, peroxisome proliferator-activated receptors PPARα and PPARγ, and transient receptor potential vannilloid-1 channels (647, 648). The study of expression of CB1Rs and mediators in sAD has yielded variable results (649). In contrast, increased CB2R expression in microglia surrounding SPs is consistently documented (650, 651). A few studies have shown altered expression levels of endocannabinoids and enzymes linked to their metabolism in sAD (652-654). Therefore, the endocannabinoid system plays a role in sAD although its precise contribution remains largely unknown. Treatment with exogenous cannabinoids and modulation of CBRs in murine models of AD has shown beneficial effects including reduction of β-amyloid plaques and β-amyloid burden, reduced tau phosphorylation, reduced inflammation, excitoxicity, mitochondrial dysfunction, and oxidative stress, and relief of cognitive impairment (649).
8. Trophic factors and receptors
The expression of trophic factors, particularly brain-derived neutrophic factor (BDNF) and nerve growth factor (NGF) and their receptors, is altered in sAD (655-659).
BDNF mRNA and protein are decreased in the frontal cortex and hippocampus in AD (660-662). BDNF immunoreactivity is reduced in tangle-bearing and non-tangle-bearing neurons, whereas immunoreactivity to full-length TrkB (the high affinity receptor for BDNF, neurotrophin-3 and neurotrophin-4) is reduced in tangle-bearing neurons. Strong BDNF immunoreactivity is observed in dystrophic neurites surrounding SPs, and strong TrkB in reactive glial cells, including those surrounding SPs. Truncated TrkB immunoreactivity occurs in individual neurons and reactive glial cells in the cerebral cortex and white matter in sAD (661). The cause of abnormal TrkB immunoreactivity in sAD is not known but β-amyloid modulates TrkB alternative transcript expression (663). ProBDNF is increased in sAD, and it is modified by reactive oxygen species (ROS)-derived advanced glycation end products, which prevent the processing of proBDNF to mature BDNF (664). Abnormal proBDNF/BDNF signaling impairs axonal transport, decreases trophic effects, and increases pathogenicity and cell death (661, 664, 665).
In contrast, the expression of NGF is increased in sAD (655, 666), but NGF-containing neurons in the basal forebrain are lost in sAD (454, 667). ProNGF is also increased in sAD (668, 669). This has relevant implications as proNGF induces processing and nuclear translocation of the intracellular domain of p75NTR (the low affinity neurotrophin receptor for NGF, BDNF, neurotrophin 3, and neurotrophin 4) and induces cell death in association with cofactor sortilin, a member of the Vps10p sorting receptor family (670-672).
9. Endoplasmic reticulum stress
The accumulation of abnormally misfolded proteins in the endoplasmic reticulum (ER) causes ER stress which is manifested by activation of one or more of the three signaling pathways of the misfolded protein response (UPR) (673). Glucose-related protein 78 (GRP78/BiP) is the master protein that regulates the UPR (674). The cytosolic domains of the transmembrane ER proteins PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor (ATF)-6, trigger specific pathways once activated. PERK activates ATF-4, and the activation of ATF6 involves its displacement from the ER to the Golgi apparatus to be cleaved into ATF6c. IRE1 dimers phosphorylate and lead to the production of X-box binding protein 1 (XBP-1) which increases the ER capacity of protein folding and the degradation of abnormally folded proteins in the ER, thus reducing ER stress. IRE1 may also bind to the TRAF2 (TNF receptor-associated factor 2) adaptor molecule and activate the apoptosis signal-regulating kinase 1 (ASK1), which in turn causes the phosphorylation of c-Jun N-terminal kinase (JNK), thereby triggering cell death. ATF-4, truncated ATF6, and XBP-1, through downstream target genes, modulate ER homeostasis or apoptosis, depending on the saturation of the system (673, 675). The expression of several UPR components is altered in aging and sAD (676-679). ER stress also generates ROS which, together with mitochondrial ROS, is a major cause of oxidative stress damage. Mitochondrial dysfunction and ER stress are relevant promoters of apoptosis in sAD (680). ER stress is also linked to brain inflammation (681, 682). Moreover, β-amyloid may activate the UPR, either mediated by glutamate receptors and calcium uptake, or linked to mitochondrial dysfunction and ROS production; ER stress may also be induced by abnormal tau (683-685). Moreover, PERK, IRE1 and ATF6 signaling pathways activate autophagy (686-688).
10. Failure to remove debris: the ubiquitin-proteasome system (UPS) and autophagy in sAD
Autophagy and UPS are the two main mechanisms of intracellular protein degradation. There is a certain relationship between these two mechanisms, and there are some molecules in common that initiate compensatory effects to prevent disease progression (689, 690).
Autophagy includes macroautophagy, microautophagy, and chaperone-mediated autophagy (691). Microautophagy and chaperone-mediated autophagy involve lysosomal membrane invagination and chaperon recognition (692). Macroautophagy is mediated by autophagosome protein assembly of Beclin 1-Vps34 lipid kinase, Atg9-WIPI-1, Atg12 conjugation system, microtubule-associated protein light chain 3 (LC3), and Unc-51-like autophagy activating kinase 1 protein kinase. Lysosomal-associated membrane protein 1 (LAMP-1) is a transmembrane glycoprotein enriched in the membrane of lysosomes (693-695). Material digested by autophagy is incorporated into lysosomes.
Autophagy is impaired in brain aging and sAD (696-706). Altered macroautophagy in sAD is manifested in dystrophic neurites of SPs, in which mitochondria, dense bodies, and vesicles are common targets, and in synapses and granulovacuolar degeneration (see section 11). Autophagy is also involved in β-amyloid metabolism and clearance (707).
UPS activity is initiated by the conjugation of ubiquitin to the substrate following a three-step cascade to tag the protein into the proteasome. The 20S proteasome is a hetero-oligomer formed by heptameric rings organized into a structure resembling a hollow cylinder which has three main peptidase activities: chymotrypsin-like, trypsin-like, and peptidylglutamyl peptide hydrolyzing activities. The 20S proteasome can associate, in the presence of ATP, with two caps or 19S complexes, thereby forming the 26S proteasome complex. The 19S complex serves in the recognition of ubiquitylated proteins, protein unfolding, and translocation of the unfolded polypeptide to the inner chambers of the 20S proteasome for hydrolysis. The 20S proteasome can interact with other complexes. The PA28α/β activator (11S regulator) can also bind to 20S proteasome to form the PA28-proteasome complex. Additionally, the three catalytic β subunits of the 20S proteasome in response to γ-interferon are replaced by inducible homologous proteins LMP2, LMP7, and MECL1, forming the immunoproteasome (689). The immunoproteasome has a role in peptide production for antigen presentation by the major histocompatibility complex in most settings, but it is also involved in the clearance of oxidatively damaged proteins (708).
The UPS is altered in brain aging and AD (709-712). Impaired removal of altered proteins is manifested by the deposition of hyper-phospohorylated, abnormally conformed, and truncated ubiquitinated tau species resistant to UPS degradation in NFTs, dystrophic neurites, and threads (197). In addition to ubiquitin, mutant ubiquitin is expressed in the aging brain and at early stages of sAD, whereas misframed ubiquitin contributes to the blockade of the proteasome by NFTs and other tau inclusions (229, 230, 713-716). Yet, mutant ubiquitin reduces Aβ plaque formation (717, 718).
The immunoproteasome is activated in AD and APP/PS1 double-transgenic mice (719-721). The reasons for immunoproteasome activation are not fully understood, but the presence of advanced glycation end products (AGEs) appears to induce activation of the immunoproteasome; the inhibition of AGE receptor (RAGE), and the downstream signalling Jak2 (Janus kinase 2)/STAT1 (signal transducer and activator of transcription 1) abolishes AGE-induced activation of the immunoproteasome (722).
11. Granulovacuolar degeneration (GVD)
GVD is characterized by the presence of vacuolar cytoplasmic lesions with a dense central core, mainly in CA1 hippocampal neurons (18). GVD is common in the hippocampus in the aging human brain and present in the majority, if not all cases, of sAD (Figure 8). GVD first appears in neurons of the hippocampal subfields CA1 and CA2, and the subiculum; this is followed by the entorhinal cortex, and CA4 neurons in stage 2, temporal neocortex in stage 3, amygdala and/or the hypothalamus in stage 4, and cingulate, frontal, and parietal cortices in stage 5 (723). GVD appears in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders, particularly AD (724). The most widely accepted model of AGD generation follows the scheme: abnormal tau in pretangles induces abnormal reticulum stress responses, which in turn trigger endocytic and autophagic pathways in the face of impaired proteolysis and altered function of the UPS. This scenario is consistent with the idea that GVD is the final stage of an active process linked to failed degradation of abnormal protein aggregates in the cytoplasm of a subpopulation of neurons (677, 725-727). Immunohistochemistry to casein kinase 1δ (CK1-δ) and AT8 and laser microdissection have identified the proteomes of neurons containing GVD and NFTs, respectively, using label-free LC-MS/MS (728). A significant change in the abundance of 115 proteins in GVD-containing neurons and 197 in NFT-containing neurons was observed compared to control neurons (728). Differences in protein composition between NFTs and neurons with GVD are further supported by the demonstration of different activation of kinases and different profiles of phosphorylated proteins (139). Nonetheless, the same study of phosphoprotein expression showed that abnormal phosphorylation of various substrates is common at the first stages of NFT and GVD generation (139). These observations suggest that GVD is not restricted to tau pathology but rather involves a varied number of proteins. Since GVD is common in the older human population, it is not surprising to find a correlation between GVD pathology and cognitive impairment. However, when analysis is controlled for other associated neuropathologies, the associations between GVD and dementia lose significance (729).
Figure 8: Granulovacuolar degeneration (black arrows) in neurons of the hippocampus. Paraffin section, p38-P immunohistochemistry, slight haematoxylin counterstaining, bar = 25 μm.
12. Glial alterations in aging and sAD
Glial cells are altered in aging and sAD. Changes in glial cells have multiple facets, including cell senescence, astrocytic gliosis, microgliosis, activated inflammatory responses, and calcium homeostasis, among others. Astrocytes are key players in AD modulating β-amyloid turnover, calcium homeosthasis, tripartite synaptic function, neuroinflammation, oxidative stress responses, and BBB dysfunction (730-734). Microglia and astrocytes, together with neurons and blood vessels, participate in the process of activation of inflammatory responses in aging and sAD (735-737). Moreover, microglia have the capacity to transform a subset of reactive astrocytes through the combination of IL-1α, TNF, and C1q (738). Both microglia and astrocytes are key participants in neuroinflammation in AD (739).
12a. Astrocytes
Astrocytes are key elements in the maintenance of the central nervous system (CNS) due to their role in brain homeostasis at all levels of organization from molecular to the whole organ (740). Astrocytes express neurotransmitter receptors, pumps, and transporters at their plasmalemma, along with transporters in the endoplasmic reticulum and mitochondria that regulate the cytosolic levels of ions which underlie most, if not all, astroglial homeostatic functions (741).
With aging, astrocytes show accumulation of lipofuscin, hypertrophy of cytoplasmic filaments, increased expression of glial fibrillary acidic protein (GFAP), S100β and vimentin, and modifications in morphology and number (742-744). Senescent astrocytes also exhibit senescence-associated secretory phenotype manifested by increased production of pro-inflammatory cytokines together with oxidative damage and increased superoxide production (745). Perivascular astrocyte senescence leads to altered BBB (746,747). This is accompanied by reduced expression of efflux transporter and increased expression of influx transporter receptors for AGEs. Abnormal transport of proteins through blood vessels affects the transfer of β-amyloid, leading to its accumulation in blood vessels (748, 749). Water aquaporin 1 (AQP1) expression in astrocytes is also altered in the frontal cortex at NFT stages I-II, suggesting early impairment of water transport linked to AD-related pathology (750). AQP4 expression is altered with aging and sAD; loss of AQP4 is associated with increased levels of β-amyloid and tau, suggesting that loss of AQP4 impairs the BBB and the gliolymphatic barrier (751).
Astrogliosis and astrocyte atrophy are relatively early events in AD (752-756). Reduced branching and reduced connexin 43 expression occur in sAD (757) which may compromise the extent of coverage domain and synaptic function in neighboring neurons (758). β-amyloid peptides also induce mitochondrial dysfunction and oxidative stress in astrocytes (759). Transcriptomics of laser-captured microdissection using GFAP as a marker revealed marked dysregulation of insulin, phosphatidylinositol 3-kinase (PI3K)/Akt, and mitogen-activated protein kinase (MAPK) signaling pathways at advanced Braak stages of the disease; minor and different abnormalities were observed at earlier stages, indicating different responses of astrocytes along disease progression (760). Recent transcriptomic studies in sAD have shown upregulation of genes related to perisynaptic astrocytic processes and downregulation of genes encoding endolysosomal and mitochondrial proteins; downregulation of astrocytic mitochondrial genes inversely correlates with the disease stages defined by Braak and CERAD scoring (761).
Reactive astrocytes are found mainly around Aβ deposits in SPs and blood vessels (752, 762, 763) and in areas without plaques then distributed in a layered pattern (762, 764). In response to vascular β-amyloid deposition, astrocytes produce cytokines, metabolizing enzymes, and ROS which in turn contribute to altered BBB and perivascular astrocytic function (765-769). Reactive astrocytes may contain Aβ (770-772) and N-terminal truncated β-amyloid (773), and they have the capacity to internalize and degrade β-amyloid fibrils (774). Activation of metalloproteinases (765, 775) and lysosomal degradation (776, 777) are the main complementary mechanisms by which astrocytes degrade β-amyloid. In addition, BACE may be expressed in astrocytes under appropriate conditions, facilitating the generation of β-amyloid in these cells (752, 778). Acquisition of a pro-inflammatory profile along with activation of the β-amyloidogenic pathway further potentiates toxicity of a subpopulation of astrocytes in AD (779). Curiously, β-amyloid seems to impair the phagocytosis of dystrophic synapses by astrocytes (780).
Astrocytes bearing β-amyloid show abnormal calcium homeostasis (758, 759, 781-783). In addition, β-amyloid-induced glutamate release by astrocytes may contribute to neuronal excitatory damage (784). De-regulation of specific metabotropic glutamate receptors in astroglia is also a putative harmful effect of β-amyloid (785).
NMDARs are expressed in a sub-population of astrocytes in the cortex and spinal cord. These receptors are composed of two GluN1, one GluN2C or D, and one GluN3 subunits. This composition makes astroglial NMDARs operational, modulating resting membrane potential (786).
EAAT2 clears excess extracellular glutamate from the synaptic cleft and extrasynaptic sites via glutamate re-uptake by glial cells and neurons to prevent neuronal hyperexcitability and excitotoxicity. Oxidative damage, splice variants, and altered solubility of EAAT2 may lead to functional alterations of glutamate transporters in AD (787-789). Moreover, EAAT2 expression is reduced with disease progression in parallel with increased GFAP expression in sAD (762, 790, 791). Furthermore, astrocytes surrounding SPs show altered immunoproteasome markers, and augmented expression of cytokines and mediators of the immune response (721, 735, 792, 793). Reactive astrocytes in sAD also have higher levels of complement component C3 which is required for both classical and alternative complement activation pathways (794).
Changes in the morphology of astrocytes are accompanied by alterations in the regulation and expression of GFAP and other astrocyte markers in cases with MCI and in pre-clinical AD (791, 795-797). These changes are preceded by expression of various cytokines and components of the inflammatory response (793, 798). Astrocytic responses are not homogeneous but rather variable, even in the same region (799-801). As for M1 and M2 suggested phenotypes for microglia (see section 12b), A1 and A2 astrocyte phenotypes have been proposed depending on their transcriptional profiles: A1 are neurotoxic and A2 neuroprotective (738, 802). However, this categorization is difficult to apply in the context of sAD (801-803).
Glucose hypometabolism is a characteristic metabolic feature associated with cognitive impairment in AD (804-807). Furthermore, neurons utilize lactate as a source of energy, and astrocytes are the main local source of neuronal lactate (808-811). Lactate levels are reduced in the brain of AD transgenic murine models (812). In the brain, the local source of ketone bodies results from fatty acid oxidation in astrocytes (813). In addition, astrocytes can produce ketones from amino acids (814). Considering that about 20% of cerebral ATP is generated from fatty acids (815), it may be inferred that impaired energy metabolism in astrocytes negatively impacts on neuronal energy metabolism, including maintenance of the energy requirements of the synapses (803, 816-818).
12b. Microglia
Microglial cells are multifunctional cells that respond to different stimuli leading to either beneficial or harmful effects depending on the production of specific molecules. Several studies have dealt with a proposed polarization of microglia into two types. M1 phenotype is stimulated by interferon-γ (IFN-γ) for the expression of pro-inflammatory cytokines, and M2 phenotype by IL-4/IL-13 for resolution of inflammation and tissue repair (819). However, the division between M1 and M2 microglial phenotypes with opposing effects in pathological conditions is probably a simplification, as complex microglial responses occur in the same setting, particularly in AD (820, 821). The term neuroinflammation, although widely used, sheds little light on the molecular consequences of microglial (and astrocytic) responses in any particular setting (822-824).
Microglia are modified in aging and AD (825-828). Dystrophic (senescent) microglia precede activated microglia in aging and sAD (826). Increased numbers of activated microglial cells occur in parallel with the production of β-amyloid and tau pathology (829). Microglial cells are found associated with SPs in contact with dystrophic neurites and internal to reactive astrocytes (7). Diffuse microgliosis involving the cerebral cortex and subcortical white matter occurs as well.
Microglial activation in AD is associated with upregulation of a large number of cytokines, chemokines, members of the complement system, and other mediators of the immune system (736, 830-837). Importantly, the inflammatory response is not homogeneous but is largely region and stage dependent (838). Inflammatory responses also occur in transgenic mouse models of AD with specific region and stage profiles. Yet, the regulation of different components of brain inflammation and the immune system differ in transgenic mice when compared with sAD (838, 839). This is an important point, as inflammatory responses vary in different regions with disease progression; protective and deleterious microglia-associated effects may occur simultaneously in any individual at any stage of the disease.
Another relevant point is the role of lipids in activated microglia; increased expression of ApoE, triggering receptor expressed on myeloid cells 2 (TREM2), and lipoprotein lipase (LP2), is found in activated microglia (840).
A link between β-amyloid and microglia is well documented (829, 841, 842). A cell surface receptor complex for fibrillary-amyloid mediates microglial activation (843). Microglia, in turn, mediates the clearance of soluble Aβ through fluid phase macropinocytosis (844). Microglia-mediated synapsis loss in AD is enhanced by fibrillar Aβ and oligomeric Aβ aggregation onto neuronal post-synaptic terminals, complement deposition, C3 receptor (CD11b/CD18) activation, microglial activation, and synapsis phagocytosis (821, 845-849).
Microglia activation also correlates with tau pathology and NFT staging (829, 850-852). Positron emission tomography (PET)-based studies have also shown a correlation between tau pathology progression and microglial activation across Braak stages (853, 854). Abnormal tau at the synapsis also favors synapsis pruning and elimination by micoglial phagocytosis (855).
The notion that two types of microglial cells, unrelated to the proposed M1 and M2 subtypes, may play different roles in sAD is supported by recent observations. Using single nuclei RNA sequencing (snRNAseq) of isolated microglial nuclei in sAD brains, the abundance of phagocytic/activated named AD1-microglia correlates with tissue β-amyloid load and localizes with β-amyloid plaques; AD2-microglia are more abundant in association with tau pathology (856).
Microglial and altered expression of inflammatory markers is observed in children with Down syndrome, and it is modified in parallel with the appearance of tau and β-amyloid pathology and through disease progression. Microglial responses in Down syndrome with AD pathology differ from those of sAD (857-859). The profile of brain inflammatory responses also differs in human cases with AD-resilient pathology; levels of trophic factors are increased, whereas expression levels of chemokines are decreased (860).
The inflammasome is a multi-protein complex containing a member of the NOD-like family, such as pyrin-domain containing 3 (NLRP3R) that interacts with the inflammasome-adaptor protein ASC. The immune response elicited by β-amyloid functions through complex crosstalk between the toll-like receptor 4 (TLR4), complement, and inflammasome signaling pathways (861). The activation of the inflammasome in microglia triggers a cascade that involves caspase 1, and maturation of several cytokines including IL-1β and IL-18. The inflammasome in microglia participates in the nucleation of β-amyloid plaques and enhances tau pathology (862, 863). Aβ aggregates, and soluble Aβ oligomers and protofibrils, activate the NLRP3 inflammasome (864-866). Conversely, activation of NLRP3-ASC inflammasome aggravates amyloid pathology (867). Aggregated tau also activates NLRP3/ASC and exacerbates tau pathology (868). Loss of NLRP3 inflammasome function reduces tau hyperphosphorylation and aggregation by regulating tau kinases and phosphatases. Conversely, tau activates the NLRP3 inflammasome. Moreover, the intracerebral injection of fibrillar β-containing brain homogenates has been shown to induce tau pathology in an NLRP3-dependent manner (869).
Microglial and inflammatory responses are dependent on genetic factors in sAD (280, 870-872). ApoE status is a modifier of the inflammatory response (873, 874). CD33, TREM2, and other genes linked with inflammation are expressed in microglia (871, 875, 876). Carriers of AD-associated risk variants in TREM2 show a reduction in plaque-associated microglia, and an increase in dystrophic neurites and overall pathological tau compared with age- and disease stage-matched sAD patients without TREM2 risk variants (877). Another study shows that TREM+ control cases have no pathological hallmarks of sAD, whereas TREM2+ sAD cases show amoeboid microglia and upregulation of inflammatory markers when compared with TREM2+ controls and TREM2- sAD cases. These findings suggest that TREM2 influences, but does not trigger, the microglial responses in sAD (878).
12c. Oligodendrocytes
Myelin generation and maintenance, and axonal nurture in the CNS, are carried by oligodendrocytes. Oligodendrocytes may contain abnormal protein deposits in various neurodegenerative diseases with abnormal protein aggregates. Yet alterations in oligodendrocytes may occur without accompanying abnormal deposits. Oligodendrogliopathy is used to stress the role of altered oligodendrocytes in the pathogenesis of certain neurological diseases with or without abnormal oligodendroglial deposits (879).
Oligodendrocytes suffer a functional decline in aging and AD as revealed by combined morphological, biochemical, and neuroradiological methods. Neuroimaging and neuropathological studies have shown reduced white matter (WM) volume, WM lesions, and altered WM integrity and cortical disconnection in aging human brain (880-884). WM changes are associated with disruption of myelin and axons (885, 886). Alterations in the number of oligodendrocytes and oligodendroglial precursor cells (OPCs/NG2-positive cells) have been reported in aged primates and rodents (887). Neuroimaging studies show reduced WM size, WM hyper-lucencies, and myelin and axon damages in patients with MCI and dementia of Alzheimer’s type (883, 888-896). WM atrophy, decreased myelin density, and demyelination are also observed in post-mortem neuropathological studies (881, 896, 897). Breakdown of WM integrity is considered a contributor to the loss of neuronal tract connectivity in aging and sAD (898, 899). Recent neuropathological studies have shown preservation of MBP, PLP1, CNP, MAG, MAL, MOG, and MOBP mRNA expression levels in the WM of the frontal cortex at NFT stages I-II/0-A when compared with MA individuals without NFT pathology, but a significant decrease at stages III-IV/0-C. This is accompanied by reduced expression of NG2 and PDGFRA (platelet-derived growth factor receptor A) mRNA, reduced numbers of NG2-, Olig2-, and HDAC2 (histone deacetylase 2)-immunoreactive cells, and reduced glucose transporter immunoreactivity at stages III-IV/0-C. Curiously, partial recovery of some of these markers occurs at stages V-VI/B-C. These changes show that myelin loss is accompanied by reduced transcription of myelin-related proteins in the WM of the frontal cortex at middle-stages of AD (342).
Early myelin loss, decreased numbers of oligodendrocytes, and region-specific alterations, followed by partial reparative responses, also occur in transgenic mouse models of AD (900-903).
13. The neurovascular system in AD
The first highlighting of the role of vascular pathology in AD was in 1989 (904). The “vascular hypothesis” of sAD has been supported more recently (905-908). Interestingly, denervation probably due to the loss of inputs from the locus coeruleus and basal forebrain was initially considered a key factor (909); altered innervation of the cerebral blood vessels linked to cholinergic deficiency is still postulated as contributing to cerebral blood flow (CBF) reduction (910, 911).
There are hundreds of papers dealing with β-amyloid angiopathy and its effects on neurovascular function, as well as the role of lipid transporters and vascular receptors in the clearing of β-amyloid in the cerebral blood vessels (912-923). Yet, there is also overwhelming evidence of altered neurovascular functioning in sAD beyond that expected in association with β-amyloid angiopathy. Neurovascular dysfunction in individuals with MCI and advanced AD is manifested by reduced CBF (cerebral hypoperfusion), reduced cerebral glucose transport, impaired BBB function, altered lymphatic function, and altered structure of the capillaries (80, 910, 924-935).
In addition to atherosclerosis and small blood vessel disease that are common in the elderly, primary involvement of arterioles and capillaries is common in brain aging and sAD. Primary blood vessel damage in sAD is characterized by atrophy and irregularities of capillaries and arterioles, edema and increased numbers of pynocytotic vesicles in endothelial cells and pericytes, atrophy of smooth muscle fibers, thickening and focal disruption of the basal membrane, increase in collagen IV, heparan sulfate, proteoglycans, and laminin in the basal membrane, increased aquaporin expression in perivascular astrocytes, and gliovascular dysfunction, among other defects (910, 936-941). Degeneration of endothelial cells is further supported by reduced staining of endothelial cell markers (942) accompanied by aberrant angiogenesis (943). Pericytes are decreased in number and show abnormal mitochondria, pinocytotic vesicles, and disorganization and accumulation of osmiophilic material in aging with cumulative damage with sAD progression (944-947). Aβ oligomers constrict human capillaries in AD by signaling to pericytes (948). Pericyte degeneration and impaired BBB function reduce Aβ clearance and increase β-amyloid accumulation in the brain. Importantly, pericyte alteration occurs at early stages of sAD and probably it has a determining role in the altered capillary permeability (949-950). It is difficult to ascertain the weight of each one of the vascular pathologies in the eventual neurovascular failure in a particular individual. Atherosclerosis, small blood vessel disease, β-amyloid angiopathy, brain hypoperfusion, altered BBB, and primary AD-linked non-amyloidotic angiopathy have cumulative effects (951, 952).
The timing and development of neurovascular failure in sAD is not a simple process. The “two-hit vascular hypothesis” suggests that early vascular damage leads to increased accumulation of Aβ deposits in the brain, which in turn provokes additional vascular damage. ApoE is one of the factors that modulate cerebrovascular integrity (953).
Impaired glucose uptake is an early event in pre-symptomatic fAD (954), and BBB breakdown is an early biomarker of human cognitive dysfunction (955), suggesting that the neurovascular system is dysfunctional at early stages of AD. Reduced expression of glucose transporter 1 (GLUT1) is manifested at early stages of sAD, and it impairs blood glucose uptake and vasculo-neuronal function (956, 957). Altered glucose transport accounts, in part, for the more complex brain glucose metabolism dysregulation in AD (958). More precise information has been obtained from the study of mouse models of cerebral β-amyloidosis mimicking β-amyloid deposits in AD (959). In the majority of these mice, cerebral blood vessels are altered and there is often impaired BBB at early stages of Aβ plaque deposition, usually preceding or in the absence of β-amyloid angiopathy (959). These observations are in line with the role of Aβ in the pathogenesis of neurovascular damage in AD (928). Unfortunately, little is known about the occurrence of different soluble, insoluble, and oligomeric amyloid species in these models.
The possible deleterious effects of abnormal tau on the integrity of the cerebral blood vessels have also been assessed (960). Tau expression is accompanied by BBB breakdown in tetracycline-regulable tau-transgenic mice; BBB function is recovered once tau levels are normalized (961). Non-structural but functional BBB dysfunction mediated by altered modulation of vasoactive factors including vasoconstrictor endothelin-1 has been suggested (962).
Altered CBF, impaired BBB, and structural anomalies in the blood vessels also occur in other neurodegenerative diseases such as Parkinson’s and Huntington’s diseases, amyotrophic lateral sclerosis, and multiple sclerosis (933, 934, 963), in which Aβ and AD-tau have no chance to play a causative role.
Neurons and astrocytes also play a role via glutamate in the regulation of CBF. Arteriole constriction depends on the metabolism of arachidonic acid (AA). Upon mGluR activation, increased AA in the plasma membrane is converted into 20-hydroxyeicosatetraenoic acid and induces vasoconstriction, or it is converted into prostaglandin 2 and produces vasodilatation (964).
Astrocytes have cardinal functions in the maintenance of BBB (see section 12a).
14. Purine and pyrimidine metabolism in sAD
Purines are heterocyclic double-ring aromatic organic molecules. Primary purines adenine and guanosine, together with one-ring primary pyrimidine nucleobases cytosine, thymidine, and uracil, are the core of DNA, RNA, nucleosides, and nucleotides. Adenosine and guanosine are purine ribonucleosides resulting from the binding of adenine or guanine to ribose, respectively. When adenine and guanine are attached to a deoxyribose ring, the resulting compounds are deoxyadenosine and deoxyguanosine, respectively. Nucleotides result from the incorporation of phosphate groups in nucleosides: adenosine monophosphate (AMP), adenosine diphosphate, adenosine triphosphate (ATP), guanosine monophosphate (GMP), guanosine diphosphate, guanosine triphosphate, and cyclic forms cAMP and cGMP are primary purine-derived nucleotides. Modified purine nucleobases hypoxantine and xanthine result from the replacement of the amino-group by a carbonyl-group from adenine and guanine, respectively, whereas methyl-guanine results from the incorporation of a methyl group to guanine. Corresponding modified purine nucleosides are inosine, xanthosine, and methyl-guanosine, respectively. Nucleotides participate in a wide variety of crucial metabolic pathways including energy metabolism and cell signaling. In addition, purine bases are incorporated to other molecules to form cofactors of several enzymatic reactions such as coenzyme A, flavin adenine dinucleotide (FAD), nicotinamide adenine dinucleotide (NADþ), nicotinamide adenine dinucleotide phosphate (NADPþ), and the corresponding reduced forms FADH2, NADH, and NADPH. S-Adenosyl methionine is made from ATP and methionine by methionine adenosyltransferase and is involved in the transfer of methyl groups to distinct substrates, including nucleic acids, proteins, lipids, and metabolites (965, 966). In addition, adenosine may act as neuromodulator on specific adenosine receptors (967) (see section 7f). Adenosine receptors also modulate the BBB (968).
Significantly decreased levels of adenosine, guanosine, hypoxanthine, and xanthine are found in the frontal cortex at stages I-II of NFT pathology, but the parietal cortex and temporal cortex show an opposing pattern at advanced stages of sAD. The activity of 5’-nucleotidase, which hydrolizes adenosine AMP to generate adenosine, is reduced in the frontal cortex mainly at NFT stages I-II, and only at NFT stages V-VI in the temporal cortex. Adenosine deaminase activity, which synthetizes inosine from adenosine, is decreased in the frontal cortex in sAD but is increased at NFT stages I-II in the temporal cortex. Finally, purine nucleoside phosphorylase activity, which metabolizes guanosine to guanine, is increased only in the temporal cortex at NFT stages I-II. Purine metabolism alterations are region- and stage-dependent and occur independently of NFTs and β-amyloid plaques (969).
In another study, 23 purine metabolism genes were analyzed with RT-PCR in the entorhinal cortex, frontal cortex area 8, and precuneus in MA individuals without NFT pathology and in cases at NFT stages I-II, III-IV, and V-VI (966). The mRNA expression levels of several enzymes were dysregulated at stages III-IV and V-VI in a region-dependent manner when compared with MA individuals. In addition, liquid chromatography mass spectrometry-based metabolomics in the entorhinal cortex identified decreased levels of xanthosine, guanine, and deoxyguanosine at stages I-II, followed by dGMP, inosine diphosphate, and glycine at NFT stages III-IV in sAD (966).
Some years ago, studies pointed to S-adenosylmethionine as a complementary candidate for therapeutic intervention in sAD (208, 970). More recently, several studies have scrutinized the modulation of purinergic signaling to ameliorate sAD (971-976). Considering the large number of different enzymes altered, this represents a tremendous endeavor.
15. Epigenetics in brain aging and sAD
Epigenetic regulation plays a crucial role in the final transcription of genes (977, 978). Three central mechanisms are briefly discussed: modifications of histones, DNA methylation and hydroxymethylation, and non-coding RNAs.
15a. Histone modifications, DNA methylation, and hydroxymethylation
Nucleosomes consist of DNA wound around a protein octamer composed of two molecules of the histones H2A, H2B, H3, and H4. The N-terminal of histones (histone tail) extends out from the surface of the nucleosome. Acetylations by histone acetyltransferases (HATs) and histone deacetylases (HDACs), and methylation mediated by histone methyltransferases (HMTs) and histone demethylases (HDMs), respectively, occur at the histone tails (979, 980). Histone acetylation decreases the interactions of histones with DNA, relaxes heterochromatin to euchromatin, and enables gene transcription (981-984). Methylation of histones can either increase or decrease gene transcription, depending on which amino acids in the histones are methylated, and on the number of methyl groups involved. Methylations, which permit DNA uncoiling facilitate the access of transcription factors and RNA polymerase. The trimethylation of histone 3 at lysine 4 (H3K4m3) activates transcription, whereas dimethylation of histone H3 at lysine 9 (H3K9me2) is inhibitory. Likewise, methylation of lysine 4 of histone 3 (H3K4me1) facilitates gene transcription (985-988).
DNA methylation and DNA hydroxymethylation are biological processes by which methyl or hydroxymethyl groups are added to the DNA. Methylation and hydroxymethylation can change the activity of a DNA segment without changing the sequence. The methylation of a 5′-cytosine in cytosine-guanine-rich regions (CpG islands) of DNA promoters results mainly in the inability of transcription factors to bind DNA and compact heterochromatin; as a result, gene transcription is silenced. Four types of DNA methyltransferases, DNMT1, DNMT2, DNMT3a, and DNMT3b, carry out the process of DNA methylation. S-adenosyl-L-methionine (SAM) is the donor of methyl groups. DNA hydroxymethylation can occur as a result of oxidative stress or the action of ten-eleven-translocation-1 (TET1) proteins (989-994). SAM which is generated by adding adenosine to methionine, a reaction catalyzed by S-adenosyl-methionine transferase, transfers the methyl group to DNA methylation by DNA methyltransferase.
Brain aging and sAD is accompanied by epigenetic DNA modifications (995-1002). DNA modifications involve genes linked to β-amyloid production (1003-1006), ApoE-ε4 (996), tau phosphorylation (1007-1009), ribosomes (1010), BDNF (1011), and inflammation (1012, 1013). A large number of unrelated genes has also been assessed, showing either changes or no modifications in the methylation of DNA promoters (1014-1023). The expression of adenosine receptor A2A is modulated by DNA methylation in the gene promoter region (1024-1026). Therefore, epigenetic changes in DNA in aging and sAD are not widespread but selective for particular genes.
In monozygotic twins discordant for AD, significantly reduced levels of DNA methylation were observed in the neuronal nuclei of temporal neocortex in the AD twin (991).
Unfortunately, most studies are carried out on samples at advanced stages of sAD which precludes learning whether epigenetic changes in DNA are primary or secondary events (1027).
In addition to modifications in genomic DNA, increased mitochondrial 5-methylcytosine in most CpG and non-CpG sites in the D-loop region of mitochondrial DNA (mtDNA) has been identified in the entorhinal cortex at NFT stages I-II and III-IV compared with control samples (1028). These are relevant data as they indicate early mtDNA methylation linked to the pathogenesis of AD-related neuropathologic change (1028).
15b. Non-coding RNAs
Non-coding RNAs do not encode proteins, but most modulate protein translation targeting mRNAs. Transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs); small non-coding (snRNAs) such as microRNAs (miRNAs), small interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), extracellular RNAs (exRNAs), small Cajal-body specific RNAs (scaRNAs); and long non-coding RNAs (lncRNAs) are the principal types. miRNAs bind to complementary un-translated regions (3’-UTRs) of mRNAs to regulate target genes, resulting in translational repression or degradation (1029-1031). In animals, miRNAs are synthesized from primary miRNAs by the action of two RNase III-type proteins: Drosha, in the nucleus, and Dicer in the cytoplasm. Argonaute (Ago) subfamily proteins bind mature miRNAs to target mRNAs (1032, 1033).
Thousands of human genes are miRNA targets (1034, 1035). Ago2:RNA interactions using HITS-CLIP have been used to generate a transcriptome-wide map of miRNA binding sites in the human brain. About 7,000 stringent Ago2 binding sites were highly enriched for conserved sequences corresponding to abundant brain miRNAs. This interactome points to functional miRNA: target pairs across about 3,000 genes (1036).
Dysregulation of miRNAs is involved in the pathogenesis of sAD (1037-1040). Several miRNAs target genes, many of them abnormally regulated in AD, are associated with β-amyloid (1041-1060), tau phosphorylation, and cytoskeleton (1061-1074). It is difficult to ascertain which miRNA alterations are primarily altered or secondary to β-amyloid and tau pathology in sAD (1075).
Several miRNAs can bind to specific mRNAs, modulating gene transcription differentially depending on the triggering factor (1038). miRNA 163, miRNA30a 5p, and miRNA206 bind to BDNF mRNA and modulate altered BDNF expression in sAD (1040, 1055, 1076).
Furthermore, one miRNA can bind to different mRNAs resulting in various functions. For example, reduced miRNA 132 is associated with downregulation of ChAT immunoreactivity in the nucleus basalis of Meynert (1061) and to AChE upregulation (1077, 1078). In addition, miRNA 132 targets CREB and promotes neuritogenesis and synaptic activity (1079).
Finally, miRNA dysregulation is stage- and region-dependent. For example, in the locus coeruleus, miRNA-27a-3p, miRNA-124-3p, and miRNA-143-3p show a trend to increase at NFT stages I-II and are significantly upregulated at NFT stages III-IV when compared with MA individuals without NFT pathology. In the entorhinal cortex of the same cases, only miRNA-143-3p is upregulated at stages III-IV. The expression levels of miRNA-27a-3p, miRNA-124-3p, and miRNA-143-3p are not modified in CA1 at any stage, whereas miRNA-124-3p is significantly downregulated in the dentate gyrus at NFT stages I-II (1080). The interpretation of these results is puzzling: downregulation of miRNA124 results in β-amyloid accumulation (1007); miRNA143-3p inhibition promotes neuronal survival in sAD (1081); and miRNA 27a-3p regulates the expression of intercellular junction proteins at the brain endothelium, while its downregulation increases BBB permeability (1082, 1083).
16. Microorganisms and sAD
The possibility that microorganisms participate in the pathogenesis of AD was suggested in a seminal publication in 1907 (1084). The role of bacteria, viruses, and fungi has been proposed to the present (1085-1088).
16a. Microorganisms in the brain and oral cavity
More than thirty years ago, Borrelia burgdorferi was reported in the brains of sAD cases (1089), although other studies did not validate this finding (1090). However, spirochetes, including Borrelia and Treponema, were postulated as contributing factors in the pathogenesis of the disease (1091-1094). rRNA sequencing has shown the presence of bacteria in Alzheimer’s post-mortem brain (1095). β-amyloid exhibits potent antimicrobial activity against Candida albicans and some bacteria, pointing to the possibility that secretion of Aβ is triggered by microbial infection (1096).
Numerous periodontal bacteria have also been implicated in sAD in the context of chronic periodontitis (1097-1099). The association between periodontal inflammatory disease and sAD has raised much concern mainly because of the possibility of rapid therapeutic intervention (1100-1102). Increased levels of anti-Helicobacter pylori-specific antibodies were also reported in the CSF and serum of sAD patients (1099).
Histological and immunohistochemical techniques have detected fungi and bacteria in the brain of sAD (1103-1105). PCR analysis revealed various fungal species in the brain and CSF in sAD (1106-1108). A combination of bacteria and fungi was observed in several cases, and several fungi were identified in different brain regions in the same individual, suggesting multi-fungal infection (1107). The corpora amylacea are particularly enriched in fungal and bacterial peptides, acting as scavengers of microbial debris (1109). The presence of fungi is not restricted to sAD as they have also been identified in the CNS and CSF in amyotrophic lateral sclerosis (1110, 1111).
Herpes viruses were proposed as participants in the pathogenesis of sAD about four decades ago (1112, 1113). Since then, several studies have implicated herpes viruses in sAD (1114- 1116). Herpes simplex virus 1 (HSV-1) has received much attention due to the presence of high expression levels in the brain and HSV-1 IgG antibodies in the CSF (1117-1121). Interestingly, HSV-1 DNA is localized in senile plaques (1122). The link between herpes zoster and sAD is still controversial (1123). Human cytomegalovirus (CMV) seropositivity was associated with a risk of sAD (1124). Human herpes virus 6 (HHV-6) has focused on several studies (1125-1128). Viral associations have also been reported, including HSV-1 and CMV, HSV-1 and HHV-6, HHV-6 and EBV (Epstein-Barr virus), and HHV-6 and HHV-7 (1115, 1126, 1129, 1130). A relationship is established between β-amyloid and herpes virus, suggesting a mechanism against brain infection (1131). Since differences in sAD and controls are not always significant, it is still difficult to conclude that a direct causal relationship exists between herpes virus and sAD.
The hypothesis of multi-pathogen infections in the pathogenesis of sAD has recently been discussed (1085). Moreover, exposure to systemic infections in 5xFAD transgenic mice, carrying mutated genes associated with fAD, causes neurodegeneration in brain regions displaying β-amyloid pathology and high local microglia density (1132).
Together, the available information shows that the human brain, particularly the aged brain, accommodates viruses, bacteria, and fungi, together with the debris of these microorganisms over time. Whether brain and oral microorganisms might contribute to “neuroinflammation” and facilitate the course of sAD remains obscure. Nonetheless, a recent review postulates a link between the β-amyloid cascade hypothesis and chronic infection in sAD: β-amyloid deposition is produced in response to brain microbial inflammation (1133).
16b. Gut microbiota
In recent years, several studies have implicated the “gut-brain-microbiota axis” in the pathogenesis of sAD; altered gut microbiota may promote neuroinflammation, Aβ aggregation, and oxidative stress (1134-1140). The putative mechanisms producing effects include changes in the systemic metabolism that may influence brain functions, transfer of microbiota metabolites to the brain through the BBB, and direct impact of microbiota reaching the brain parenchyma. Serotonin may play a role in the gut-microbiota axis (1141). The opioid system has been postulated to be contributory as well (1142).
Conversely, the infusion of tracers into the lateral ventricle of rats has permitted their visualization in the nasal cavity, nasal pharynx, soft palate, and esophagus, thus suggesting that at least rats can swallow waste from the brain (1143).
17. Seeding and spreading of β-amyloid and tau
17a. Seeding β-amyloid
The intracerebral injection of diluted extracts from AD brains or from old AD-like transgenic mice accelerates β-amyloid deposition in transgenic mice bearing APP and/or PSEN1 mutations (1144-1148). Soluble forms of Aβ are particularly effective at inducing plaque formation (1149). Plaques not only develop locally but extend as well to the cerebral cortex distant from the injection site. These findings support the possibility that once the first β-amyloid deposits appear they may accelerate the accumulation of additional seeding in other parts of the brain, thereby contributing to the exponential deposition of β-amyloid (1150). Neuronal activity regulates the regional vulnerability to β-amyloid deposition (1151).
β-amyloid seeding and spreading may also occur following peripheral inoculation of β-amyloid seeds (1152). However, the intracellular mechanisms linked to the formation and fibrillisation of host β-amyloid and recruited β-amyloid spreading need clarification.
Intraventricular injection of Aβ oligomers in cynomolgus macaques leads to diffusion into the brain, causing tau hyperphosphorylation, NFT formation, synaptic loss, and astrocyte and microglial activation in regions of the macaque brain where Aβ oligomers are abundant (1153).
β-amyloid deposits were also observed in the brains of patients with iatrogenic Creutzfeldt-Jabob disease (iCJD) secondary to cadaveric dura mater grafts, treatment with cadaveric human growth hormone obtained from hypophysis of CJD-affected donors, or contaminated neurosurgery early in life (1154).
17b. Tau seeding
Physiological release of neuronal tau is stimulated by neuronal activity and extracellular tau in vivo (1155-1158). Intercellular tau transmission may have physiological functions that permit the transfer of tau status information from one neuron to another, enabling them to modify the status of the host tau according to this information. This mechanism may lie behind the activation of post-translational modifications of tau in the host neuron following the transfer of pathological tau in models of tau seeding and spreading. Tau is secreted by exosomes, and the extracellular appearance of tau does not depend on neuronal death, but rather physiological tau transfer from one neuron to another (1159). Tau phosphorylation facilitates tau transmission and propagation (1160).
Several studies have suggested that the progression of sAD and other neurodegenerative diseases with abnormal protein aggregates occurs in a similar way to prions in prion diseases (1161-1164).
In favor of this hypothesis, many experimental designs in mouse and rat models have provided evidence that cells have the capacity to transfer tau from one cell to another. The over-expression of human tau P301L restricted to the entorhinal cortex shows the progression of tau without detectable transgene expression anterogradely from the entorhinal cortex to the dentate gyrus and CA1 region of the hippocampus and subiculum, and retrogradely to scattered neurons in the perirhinal and secondary somatosensory cortex (1165, 1166). Using a lentiviral-mediated rat model, tau protein is axonally transferred from hippocampus neurons to neurons of distant brain regions such as the olfactory and limbic systems (1167)
Several mechanisms of tau release and uptake have been proposed, mainly in the context of synaptic transmission. However other processes may occur as well. Tau release may occur via (SNAP: Soluble NSF Attachment Protein Receptor) SNARE-mediated exocytosis, release by secretory vesicles from lysosomes, microvesicle shedding, and direct plasma membrane crossing. Tau uptake may occur via endocytosis, adsorptive endocytosis, macropinocytosis, transmembrane diffusion, and nanotunneling. Binding of phosphorylated tau to AMPA and NMDA receptors is another putative mechanism (1168-1179).
Tau seeding and spreading is produced following the intracerebral inoculation of synthetic tau fibrils (1180, 1181), and the inoculation of fibrillar-enriched fractions from human and mouse brain homogenates of tauopathies, including sAD, containing hyper-phosphorylated tau (1182-1186). Tau seeding and spreading in neurons also occur following intracerebral inoculation of similar tau aggregates in wild type mice (WT) and in transgenic WT mice (1183, 1187-1194). Propagation in these models occurs through connectivity rather than proximity (1182). In addition to neurons, deposits may also occur in astrocytes, and the morphology of glial inclusions appears to mimic the glial aggregates of the corresponding human tauopathies (1183, 1188, 1189, 1195). Differences in the type of inoculum that produce different protein aggregate deposits in the host and variable involvement of astrocytes have led to the proposal that tau strains are behind the different phenotypes and progression of human tauopathies (1161, 1196-1198).
More refined situations probably occur in human neurodegenerative diseases. For example, sAD brain contains different tau strains with particular properties (1199); different tau strains may contribute to the clinical heterogeneity of AD (1200).
Moreover, tau seeding and spreading also occur in oligodendrocytes in WT mice following inoculation of sarkosyl-insoluble fractions in the hippocampus and corpus callosum of human brain homogenates from sAD and cases with primary age-related tauopathy (PART) (1191-1193). Yet, tau deposits in oligodendrocytes never occur in humans affected by sAD and PART without co-morbidities (see section 21 for PART details). These discrepancies between human diseases and mouse models may be explained by the differences between human and murine tau (1201, 1202). Whether different types of tau occur in neurons and glial cells, or even in different neuron populations granting selective vulnerability, deserves further research.
Tau seeding and spreading have also been generated in rhesus monkeys after the inoculation of adeno-associated virus expressing a double-4Rtau mutation in the left hemisphere (1203). Tau spreading was accompanied by robust TREM2+ microglial proliferation (1203), an unexpected observation not seen in other models.
Another curious observation that deserves validation is the triggering of tau deposits in tau transgenic mice following peripheral administration of tau aggregates (1204).
Neuropathological studies in humans reveal the beginning of tau seeding in the telencephalon in sAD in the transentorhinal and entorhinal cortex, and from these brain regions tau spreads to others (1205, 1206). Neuroimaging analysis further support tau spreading along functional connectivity networks (1207). However, combined post-mortem regional seed amplification and PET studies in living individuals suggest that from NFT Braak stage III onward, local replication rather than spreading between regions is the main mechanism to explain progressive tau burden in sAD (1208).
Together, tau seeding and spreading differ from seeding and spreading of prions. Grossly, tau seeding and spreading circumvent certain regions that should be involved according to the hypotheses of either neuronal connectivity or neuronal proximity. Most importantly, once up-taken by the receptive neuron, abnormal tau triggers a series of molecular processes that include activation of several tau kinases, post-translational modifications of tau—for instance, hyperphosphorylation, nitration, and altered conformation; recruitment of additional substrates in tau deposits; and modifications in the ratio of 3Rtau/4Rtau in the host neuron (1209), which implies the capacity of foreign abnormal tau to modulate exon 10 splicing in MAPT in the host (1190, 1191, 1202, 1209).
17c. Multiple seeding foci of β-amyloid and tau pathology; vulnerable and resistant populations to tau seeding in brain aging and sAD
Another important point is the characteristics of tau spreading in the human brain that seem to skip over obligate regions on the basis of cell connectivity. The dentate gyrus does not show tau pathology in AD and other 3R+4R tauopathies without co-morbidities, but the dentate gyrus is affected in 3R-tauopathies and 4R-tauopathies. Alternative pathways from the entorhinal cortex to the hippocampus proper are possible but there is no clear explanation for this particular pattern in AD. It may be suggested that certain populations are able to transfer abnormal tau from one neuron to the next in the connecting pathway without recruiting host tau and forming local aggregates within themselves. Regional vulnerability plays a cardinal role in tau spreading in AD-related animal models (1210).
In summary, tau seeding and spreading is not the only cause of tau progression in sAD and other tauopathies. Cellular vulnerability, including neuronal, astroglial, and oligodendroglial, together with regional vulnerability, already highlighted several years ago, are also key points to understanding tau progression in sAD and other tauopathies.
Finally, several studies implicate microglia in the process of tau seeding with apparent enhancing and mitigating effects (868, 1211-1216). Another study concludes that microglia have a complex role; they are capable of taking up and breaking down seed-competent tau, but do so inefficiently and could hardly play a role in the spread of tau pathology (1217).
The capacity for seeding of β-amyloid and tau does not imply a unique origin of β-amyloid and tau pathology in sAD. Neuropathological studies at early stages of NFT and SP pathology have shown that independent tau deposits are localized in separate regions such as the raphe nuclei and locus ceruleus, transentorhinal and entorhinal cortex, and olfactory bulb, in addition to isolated tau-positive neurons in other parts of the brain. Similarly, β-amyloid deposits in the brain parenchyma are not connected; likewise there is not an intimate connection of blood vessels affected by β-amyloid angiopathy.
Aβ and tau pathology, like other molecular alterations in brain aging and sAD, have multiple and separate foci; lesions in aging and sAD do not start in a single region only to progress from that site to the whole brain.
18. Neuronal death
Neuronal depletion in AD is the result of cell death and lack of neuronal renewal. Apoptosis was postulated as a major mechanism of neuronal death in AD and most neurodegenerative diseases based on the positivity of cells stained with the method of in situ end-labeling of nuclear fragmentation, and immunoreactivity to cleaved caspase 3 (1218-1220). Caspase 3-mediated and caspase 3-non-mediated apoptosis is also supported by studies in cell lines (1221-1224). However, DNA fragmentation occurs during the agonic state and post-mortem delay, and active caspase-3 participates in tau truncation during the genesis of NFTs. Therefore, the weight of apoptosis as a cause of neuron demise in AD is probably unevenly represented. Necroptosis is another type of programmed cell death, frequently linked to inflammation that depends on the kinase activity of receptor interacting protein 3 (RIP3) and RIP1, and the subsequent activation of the mixed lineage kinase domain-like protein (MLKL). This pathway is activated in AD and other neurodegenerative diseases (1225-1227).
Neuronal degeneration and demise in AD is not correlated with tau pathology (1228, 1229), although ghost tangles are not rare in advanced stages of AD. Intraneuronal accumulation of β-amyloid is involved in synaptic dysfunction, cognitive impairment, and the formation of amyloid plaques in AD (416, 1230, 1231). In addition, excessive production of Aβ peptides and APP activates death signaling pathways including apoptosis, necrosis, necroptosis, oxytosis (GSH-mediated oxidative cell death), pyroptosis (inflammasome-mediated cell death), and autophagy in vitro (1232).
In summary, although β-amyloid and tau pathology may induce cell death in sAD, they are not the only factors triggering active cell death pathways. Other alterations, such as aberrant cell-cycle re-entry, energy metabolism failure, oxidative stress damage, and altered cell membrane signaling, are relevant contributory factors (see section 21).
19. Neuronal connectivity networks in brain aging and sAD
The notion that clinical manifestations in brain aging may correspond solely to early stages of AD-related pathology restricted to the hippocampus, inner regions of the temporal cortex and selected nuclei of the brain stem is simplistic, and probably untrue. Functional neuroimaging techniques have shown selective modifications of the intrinsic connectivity network in aging that exceed the domains of these regions. The default mode, salience, dorsal attention, fronto-parietal control, and auditory, visual and motor networks decline by middle adulthood, but the motor network shows increased connectivity in middle adulthood, followed by a lessening (1233-1237). Disruption of neuronal connectivity networks appears in aging and is intensified in AD (1237-1243).
However, functional connectivity alterations do not always correlate locally to tau and β-amyloid deposition; hyper- and hypo-connectivity cycles can occur repeatedly at different stages of the disease (1244). Moreover, greater segregation of functional connections into distinct large-scale networks is associated with cognitive resilience at early stages of sAD (1245, 1246). Additional studies suggest a compensation phase followed by a degenerative phase in aging, and in early, preclinical AD (1247). This is important information, as plasticity changes may circumvent, at least temporarily, deficient functioning during brain aging and early stages of sAD. Plasticity may explain the particular capacities of the adult brain to manage information in creating new networks, manifested as “experience”.
Considering the areas affected in functional neuroimaging studies, it may be inferred that molecular changes take place in the aging brain in regions other than those affected by tau and β-amyloid pathology.
20. Human brain aging and preclinical AD
The combination of clinical manifestations and complementary biomarkers has established consensus criteria to define different clinical phases of AD (See Box 1). At preclinical stages, individuals may have measurable brain changes that indicate the earliest signs of AD (biomarkers), but they have not yet developed symptoms such as memory loss (https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf). The preclinical phase is further categorized into three stages. Preclinical AD stage 1 includes cognitively normal individuals with abnormal β-amyloid markers and no neurodegeneration; stage 2, individuals with abnormal β-amyloid and neurodegeneration as revealed by magnetic resonance imaging (MRI); and stage 3, individuals with abnormal β-amyloid deposition and neurodegeneration, and “subtle” cognitive changes (2, 48, 1248-1252). The refinement of the diagnosis of preclinical stages has improved with the acquisition of new neuroradiological methods, and optimization of CSF and blood biomarkers (1253-1256).
However, improvement in detection using CSF, blood, and plasma biomarkers largely depends on the availability of optimized instruments and markers. Current CSF and plasma biomarkers used in the diagnosis of AD are levels of β-amyloid, phospho-tau, tau, and phospho-tau ratio, neurofilaments, synaptic proteins, markers of activated astrocytes, and inflammatory markers. At present, the available methods cannot detect differential levels of tau, phospho-tau, β-amyloid, and structural and synaptic proteins unless the degenerative process is at least at undetermined middle stages of AD neuropathological change. Hippocampal atrophy, as revealed by computed tomography (CT) and MRI, and currently used to correlate cognitive impairment and altered neuropathological substrate, is a late marker of AD that is only positive when there is advanced NFT pathology and neuron loss in the hippocampus.
A recent meta-analysis to estimate the prevalence of Aβ pathology as measured with biomarkers in participants with normal cognition, subjective cognitive impairment, or mild cognitive impairment revealed that about 25-35% of cognitively normal older adults harbored a significant amount of β-amyloid (1257). Further studies have shown that β-amyloid CSF-based estimates using adjusted data-driven cutoffs are up to 10% higher than PET-based estimates in people without dementia thus suggesting that preclinical AD may be more prevalent than previously suspected (1258).
In contrast, tau PET imaging using optimized tracers provides more precise information in vivo at the different stages of NFT progression (855, 1259-1267). Improved neuroimaging methods corroborate earlier neuropathological observations in post-mortem brains. Individuals without cognitive impairment may have tau or β-amyloid positive signal alone, or β-amyloid and tau pathology, or tau and β-amyloid PET negativity (1268-1270). Neuroimaging studies have also identified tau pathology in the entorhinal region and mesial temporal cortex in normal aged individuals with or without minimal cognitive impairment (1271-1276). Tau PET observations are also useful to show that tau deposits appear before β-amyloid deposits in most individuals. Finally, neuroimaging allows the visualization of a continuum of lesions from “normal” brain aging to Alzheimer’s type dementia in sAD and fAD (1277, 1278).
Therefore, neuroimaging observations further suggest that tau pathology is predominant in normal brain aging that overlaps with tau pathology in individuals with preclinical AD. Clinical criteria of AD are biased by the consideration that Aβ pathology precedes tau pathology in sAD. In other words, the conception of the β-amyloid cascade hypothesis permeates the diagnosis of sAD.
In addition to clinical and biomarker criteria, new neuropathological standards, based on the study of a wide number of selected brain regions stained with selected immunohistochemical markers, take into consideration AD pathology progression and the presence of comorbid pathologies were proposed by the National Institute on Aging–Alzheimer’s Association (NIA-AA) in 2012 (49, 50). The main considerations are a) recognition that AD neuropathologic changes may occur in the apparent absence of cognitive impairment, (b) consideration of an “ABC” score for AD neuropathologic change that incorporates histopathologic assessments of β-amyloid deposits (named A, based on Thal phases), staging of neurofibrillary tangles (named B, based on Braak stages), and scoring of neuritic plaques (named C, based on CERAD), and (c) assessment of co-morbid conditions such as Lewy body disease, vascular brain injury, hippocampal sclerosis, and TDP-43 proteinopathy; also including ARTAG and AGD. At present, the post-mortem neuropathological examination is still the most precise approach to identify the morphological changes linked to a brain aging and sAD (49, 50, 325) (Table 1).
“A” Thal phase for Aβ plaques |
“B” Braak and Braak NFT stage |
“C” CERAD neuritic plaque score |
0 |
0 |
0 |
none |
0 |
none |
1 |
1 or 2 |
1 |
I or II |
1 |
sparse |
2 |
3 |
2 |
III or IV |
2 |
moderate |
3 |
4 or 5 |
3 |
V or VI |
3 |
frequent |
Table 1: “ABC” score for AD neuropathologic change modified from ref. 49 and 50. National Institute on Aging–Alzheimer’s Association (NIA-AA) guidelines for the neuropathologic assessment of Alzheimer’s disease.
Following this classification, the score levels of AD neuropathologic change are classified as “not”, “low”, “intermediate”, and “high” (Table 2). The presence of Aβ is mandatory. It is also stated that “medial temporal lobe NFTs in the absence of significant Aβ or neuritic plaques occurs in older people and may be seen in individuals without cognitive impairment, with mild impairment, or with cognitive impairment from causes other than AD” (49, 50). To support this assertion, a previous work is cited: “Brains that have many NFTs in medial temporal lobe structures (Braak stage III or IV) but no cortical SPs may be a diagnostic dilemma; they also raise questions about the amyloid cascade hypothesis of AD in which NFT development is thought to occur downstream of the development of amyloid plaques” (1279). More recently, the introduction of the term PART has been used to name cases with NFT pathology without SPs (see section 21a).
AD neuropathologic change |
B |
A |
C |
0 or 1 |
2 |
3 |
0 |
0 |
not |
not |
not |
1 |
0 or 1 |
low |
low |
low |
2 or 3 |
low |
intermediate |
intermediate |
2 |
Any C |
low |
intermediate |
intermediate |
3 |
0 or 1 |
low |
intermediate |
intermediate |
2 or 3 |
low |
intermediate |
high |
Table 2: “ABC” score level of AD neuropathologic change. Aβ/amyloid plaques (A), NFT stage (B), and neuritic plaque score (C). The combination of A, B, and C scores is designated as “not”, “low”, “intermediate” or “high” AD neuropathologic change. “Intermediate” or “high” AD neuropathologic change is considered sufficient for dementia. National Institute on Aging–Alzheimer’s Association (NIA-AA) guidelines for the neuropathologic assessment of Alzheimer’s disease.
A key point of this neuropathologic classification is the notion that β-amyloid deposition is the first “sine-qua-non” condition to consider the possibility of AD. The occurrence of NFTs without β-amyloid plaques is not consistent with AD at any stage. The neuropathological proposal of the National Institute on Aging-Alzheimer’s Association assumes the “amyloid cascade hypothesis” as the cause of sAD. Yet the cognitive status correlates with NFT burden rather than with β-amyloid plaques (331).
21. Primary age-related tauopathy (PART), rapidly progressive sAD, and sAD resilience
21a. PART
In 2014, the term PART, a common pathology associated with human aging, was proposed to categorize a subpopulation of individuals with normal cognition or with MCI showing NFT pathology at stages I-IV of Braak and no SPs, and slower progression to the clinical stage of dementia in some individuals (1280-1283). The concept of PART derives from the interpretation of the “β-amyloid cascade hypothesis” as the cause of AD. In other words, it is assumed that the presence of NFTs without Aβ deposition is not AD and therefore, cases with NFTs and without SPs do not match with the neuropathologic assessment of AD proposed by the NIA-AA.
PART includes the majority of individuals aged 65 or older with first stages of NFT pathology and without SPs. The individuals represent about 85% of human beings at the age of 65 (328, 329, 1284), and are considered clinically affected by “normal brain aging”. Therefore, PART appears to be a predominant disorder until the emergence of SPs (41, 329, 336, 1284). Once a substantial number of SPs appears in the brain, AD is overriding, whereas dementia with only tangles (or tangle-predominant dementia) which would be the logical progression of PART decays and becomes rare (1285). It has been stated that the frequency of PART increases at the age of 85 years (1286). However, most individuals with NFT pathology at stages I-III without SPs are younger in largest series of cases (41, 329, 1284).
The distribution of tau pathology in CA1 and CA3 regions of the hippocampus is reported to be different in PART and sAD. However, in one series, the mean age of the individuals was about 88 but the NFT stages varied in the samples with PART and sAD (1287). In another series (mean age 84.3 ± 9.4 years), tau pathology was high in both CA1 and subiculum, followed by CA2/3, entorhinal cortex, CA4, and dentate gyrus in sAD. In PART, the severity of tau pathology in CA1 and subiculum was high, followed by enthorinal cortex, CA2/3, CA4, and dentate gyrus (1288). Differences appear to be minimal and were probably modified by the presence of tau pathology in SPs in sAD.
The proposal of PART as a new tauopathy is not widely accepted. PART has also been interpreted as being within the spectrum of sAD (a part of sAD) in cases with a particular genetic background characterized by lower prevalence of ApoEε4, PTK2B, BIN1, and CR1 genes, and higher prevalence of ApoEε2 (1282, 1289, 1290). In contrast, tangle-predominant dementia has been associated with MAPT H1 haplotype (1291).
21b. Rapidly progressive AD
Patients with rapidly progressive AD (rpAD) are younger and have a median survival time after diagnosis of about 7-10 months (1292); the neuropathological hallmarks and peripheral biomarkers are similar to those seen in current sAD excepting for a low frequency of ApoEε4 allele and increased serum levels of specific pro-inflammatory cytokines (1293, 1294). Yet Aβ42 oligomers in rpAD have distinct properties which promote the faster spread of Aβ42 pathology (1295). The composition of SPs also differs in rpAD, with significantly higher levels of neuronal proteins, decreased levels of astrocytic proteins, and particular abundance of synaptic-derived proteins (1296). Moreover, high-density oligomers of the prion protein and a significant 1.2-fold decrease in di-glycosylated PrP isoforms occur in rpAD. Fifteen proteins appear to interact with PrPC while only two proteins, 3/4histone H2B-type1-B and zinc alpha-2 protein3/4, are specifically bound with the PrP isoform isolated from rpAD cases (1297). Abnormal PrP isoforms in rpAD are accompanied by altered localization of distinct interactors including the growth arrest-specific 2-like protein and associated end-binding protein 1, α-tubulin, and β-actin (1298).
Molecular profiles of β-amyloid proteoforms differ in rpAD when compared with typical sAD (1299). Moreover, the presence of highly hydrophobic Aβ seeds in rpAD brains that seeded reactions at a slower pace in comparison to typical sAD has been validated (1299).
Another anomaly is the downregulation and dislocalization from the nucleus to the cytoplasm of the splicing factor proline and glutamine rich (SFPQ), its colocalization with TIA-1 in stress granules, and its association with tau oligomers in the brain of rpAD (1300).
21c. sAD resilience
Centenarians have resistance to sAD or very low progression to advanced Braak stages of tau and β-amyloid pathology (1301-1303). Curiously, AD-related changes in the oldest-old population also show a particular neuropathological distribution including high densities of NFTs, mainly in the hippocampus, without apparent major clinical deficiencies (1302-1307).
Multiple factors are associated with increased or reduced structural and behavioral patterns linked to cognition in the elderly and sAD. The cognitive reserve, linked to educational and occupational acquisition, social networks, and leisure activities in later life, have a protective effect (1308-1312). Glycolitic dysfunction reduces resilience (1313) whereas coffee and cacao favour protection from sAD (1314). Comorbidities, including TDP-43 proteinopathy and mesial sclerosis, reduce resilience (1315-1317).
Genetic factors also participate in cognitive resilience (1318, 1319). A greater number of genes apply in women (225, 1320). A top variant on chromosome 18 upstream of ATP8B1 is significantly associated with methylation in prefrontal cortex tissue at multiple CpG sites, including one just upstream of ATPB81, and present in individuals with unimpaired cognition (1321). A rare variant in the 3'-UTR of RAB10 is protective for sAD (1322). HDAC4, REST, and Gi intracellular signaling are sAD-specific pathways involved in regulating the onset of memory deficits (1323). Molecular modulators of neuronal vulnerability, such as RAR related orphan receptor B (RORB), and glial dysfunctions are also involved in neuronal vulnerability (1324). The myocyte-specific enhancer factor 2C (MEF2C) is upregulated in a subpopulation of glutamatergic neurons in resilient individuals. Over-expression of Mef2a/c in the PS19 transgenic mouse model of tauopathy improves cognitive flexibility and reduces hyperexcitability (1325). The profiles of brain inflammatory responses also differ in resilient sAD cases, with expression levels of chemokines decreasing and trophic factors increasing (860, 1326). Yet a consensus is needed on how to define the concepts of cognitive reserve, resilience, and resistance linked to cognition in aged individuals (1327).
The genetic and molecular factors linked to the AD spectrum are listed in Table 3.
FAD |
mutations in the genes APP, PSEN1 (presenilin1), and PSEN2; increased APP dosage |
sAD |
ε4 of ApoE, LRP1, LDLR, interleukin 1a, CLU, PICALM, CR1, BIN1, TREM2, SORL1, ADAM10, ABCA7, SPI1, PILRA, MSA4, CD2AP, and EPHA1 |
PART |
lower prevalence of ApoEε4, PTK2B, BIN1, and CR1 genes, and higher prevalence of ApoEε2 |
rpAD |
low frequency of ApoEε4 allele; increased inflammation; different Aβ oligomers; different amyloid‑β proteoforms; different seeding capacities of β-amyloid; high-density PrP oligomers; decreased PrP di-glycosylated isoforms; specific PrP isoform; altered localization of the growth arrest-specific 2-like 2 protein (G2L2), α-tubulin and β-actin; downregulation and dislocalization from the nucleus to the cytoplasm of SFPQ, their colocalization with TIA-1 in stress granules, and their association with tau oligomers |
resilient AD |
variant of chromosome 18 upstream of ATP8B1; rare variant in the 3'-UTR of RAB10; MEF2C upregulation in a subpopulation of glutamatergic neurons; decreased expression levels of chemokines and increased levels of trophic factors |
Tangle-predominant dementia |
association with MAPT H1 haplotype |
Table 3: Genetic and molecular factors linked to the AD spectrum (see sections 3, 4 and 21 for references).
Considering the previous data, and the preponderance of tau pathology over β-amyloid deposits at the first and middle stages of AD-related pathology (344), a modification to the NIA-AA “ABC” score level of AD neuropathologic is hereby proposed. The added value is the reflection of tau pathology at the same diagnostic value as β-amyloid (Table 4).
AD neuropathologic change |
A |
C |
B |
0 |
0 |
0 or 1 |
2 |
3 |
0 |
0 |
low |
low |
intermediate |
1 |
0 or 1 |
low |
low |
intermediate |
2 or 3 |
low |
intermediate |
intermediate |
2 |
Any C |
low |
intermediate |
intermediate |
3 |
0 or 1 |
low |
intermediate |
intermediate |
2 or 3 |
low |
intermediate |
high |
Table 4: Modified “ABC” score level of AD neuropathologic change. Aβ/amyloid plaques (A), NFT stage (B), and neuritic plaque score (C). The combination of A, B, and C scores is designated as “not”, “low”, “intermediate” or “high” AD neuropathologic change. “Intermediate” or “high” AD neuropathologic change is considered sufficient for dementia. Here, NFTs are considered with equal relevance to SPs in the progression from normal brain aging with NFT changes, PART, preclinical AD, and sAD (including AD variants, rpAD and ADNC in centenarians).
22. Biochemical changes beyond tau and β-amyloid at the the first stages of NFT pathology
In addition to the classical neuropathological hallmarks restricted to the inner region of the temporal lobe and selected nuclei of the brain stem which characterize the first stages of NFT pathology, there is cumulative evidence of molecular and biochemical alterations in other regions in the same brains. Several brain regions such as the frontal cortex, which does not show NFT pathology at stages I-III (and very rarely, if present, β-amyloid deposition at these stages), show altered lipid and protein membrane composition, abnormal mitochondrial function, oxidative stress damage, altered protein synthesis, activation of kinases, dysregulated protein phosphorylation, and abnormal inflammatory responses, among other disruptions. All of these alterations advance in parallel with the progression of NFT pathology; most of them have implications in tau and β-amyloid pathology, and they all persist or increase at middle and advanced NFT stages and sAD. The terms NFT and sAD in these sections are those used in the original publications.
22a. Aberrant cell-cycle re-entry, and altered adult neurogenesis
The nervous system has plastic capacities and this is manifested in many ways in sAD. One of these is the activation of pathways geared to activate cell cycle re-entry. However, neurons are post-mitotic cells that activate cell death programs at G1/A and G2/M points in response to cell cycle reactivation (1328). Aberrant neuronal cell-cycle re-entry, as revealed by the expression of various proteins involved in the activation and progression of the cell cycle, is produced in a subpopulation of neurons in sAD (1329-1340). The expression of cell-cycle-related proteins occurs before the appearance of NFTs and SPs and may trigger programmed cell death or the activation of kinases leading to oxidative stress damage, tau hyperphosphorylation, and activation of β-amyloid pathways (1341-1346).
The expression of p75NTR in doublecortin (DCX)-immunoreactive dentate gyrus progenitors is reduced in AD and related transgenic models. The inoculation of pro-NGF neutralizing antibodies into the dentate gyrus restores memory performance of APP/PS1 animals and significantly increases the percentage of DCX+ progenitors in the dentate gyrus of these animals, thus suggesting that impaired proNGF-p75NTR signaling blocks adult neurogenesis in AD (1347). Moreover, the proNGF/p75NTR signaling pathway blocks adult neurogenesis and neuron cell death in AD (670-672).
Altered adult neurogenesis is not restricted to sAD, as it occurs as well in the dentate gyrus in other neurodegenerative diseases such as Huntington’s disease, amyotrophic lateral sclerosis, Lewy body disease, and frontotemporal dementia (1348).
22b. Brain lipids
The brain is the second richest organ in lipids after the adipose tissue. Cholesterol accounts for 20-25% of the total lipids in the plasma membrane of neurons; most local cholesterol is synthesized by astrocytes in the adult brain (1349). Glycerophospholipids are the main phospholipid components ubiquitously found in cell membranes, and they are found in abundance in membranes from neural cells. Lipids carry out, in addition to structural functions, the roles of mediators or second messengers. These are lipophilic molecules involved in signal transduction processes. Lipid mediators are derived from the enzymatic degradation of glycerosphingolipids, sphingolipids, and cholesterol by phospholipases, sphingomyelinases, and cytochrome P50 hydroxylases, respectively. Eicosanoids such as prostaglandins, leukotrienes, and lipoxins are derived from oxidation of the AA. Docosanoids, including neuroprotectins, resolvins, and maresin, are mediators of docosahexanoic acid (DHA). These mediators are important modulators of oxidative damage, inflammation, and apoptosis. Other glycerophospholipid-derived lipid mediators are diacylglycerols (DAGs) phosphatidylinositol 1,4,5-triphosphates, platelet-activating factor, lysophosphatidic acid, and endocannabinoids (1350). Degradation of sphingolipids also results in the generation of mediators, such as ceramide, ceramide 1-phosphate, sphingosine, and sphingosine 1-phosphate. These mediators are involved in differentiation, growth, cell migration, and apoptosis. Cholesterol-derived lipid mediators, including 24- and 25-hydroxycholesterol, produce apoptosis (1351-1354).
The physical-chemical properties of the membrane bilayer and the chemical reactivity of fatty acids determine their susceptibility to oxidative damage (1355-1357). ROS and reactive nitrogen species (RNS) are more soluble in the fluid lipid bilayer than in the aqueous solution (1358, 1359). More importantly, polyunsaturated fatty acid (PUFA) residues of phospholipids are very sensitive to oxidation (1360). As a result, neural cell membrane lipids become primary targets of oxidative damage and lipid peroxidation.
In the aging brain, there is a progressive decrease in the levels of cholesterol, phosphatidylethanolamine, phosphatidyl inositol, phospholipid, ethanolamine plasmalogen, and sphingomyelin (1361-1366). Increasing age is associated with progressive modifications in the composition of PUFA, including DHA and AA levels (1367). The age-related reduction in PUFAs is inversely correlated with stearoyl-CoA desaturase expression and activity, resulting in higher levels of monounsaturated fatty acids (MUFAs).
Lipofuscin located in secondary lysosomes increases with age in neurons and glial cells (1368, 1369). The “aging pigment” is composed of two-thirds protein and one third lipids (1370, 1371). Proteins in lipofuscin belong to the cytoskeleton, mitochondrial bioenergetics, synapse, and membrane receptors (1372). This proteome is practically identical to the proteome derived from lipoxidation reactions identified in the frontal cortex of aged humans (1373).
Levels of glycerophospholipids, sphingolipids, DAGs, and plasmalogens are altered in the brain in sAD (1374-1379). The disturbance of human brain lipid content in sAD pathology may be categorized into four main groups (i) decreased expression of phospholipids, specifically plasmalogen PE and plasmalogen PC, due in part to abnormal peroxisome activity; (ii) reduced sulfatide content; iii) increased levels of ceramides; and (iv) increased lipoxidative stress (1354). Plasmalogen PE plays a particular role as anti-oxidant (1380). Moreover, in gray matter, the major PPE molecular species are enriched in DHA and AA.
Modifications in DHA, AA, and PUFAs in sAD produce an imbalance between their protective role (the adaptive responses derived from their lipid mediators) and a deleterious role (derived from their susceptibility to oxidation) (1381, 1382). Lipid peroxidation is an early event and a major cause of oxidative stress damage in sAD progression (1373, 1381). β-amyloid plays an important part since SPs are always surrounded by oxidized lipids, as revealed using Fourier transform infrared microscopy (1383). One mechanism to modulate oxidative damage is mediation by the upregulation of DHA synthesis, which occurs in selected regions at early stages of sAD; this is followed by an adaptation, and then decreased DHA contents. This change is not uniform, but rather region- and stage-dependent. DHA content in sAD progression is like a shock wave manifested in three steps: upregulation, adaptation, and depletion.
Crucial information connecting lipid alterations and sAD comes from genetic data. In addition to allele ε4 of ApoE (268-271, 1384, 1385), many genes involved in sAD are linked to cholesterol and lipid metabolism (284, 1386-1391). Most of them increase the risk of β-amyloid deposition but ApoEε2 allele appears to decrease it (1392, 1393).
22c. Lipid rafts and cell membranes
Lipid rafts are microdomains of cell membranes enriched in glycosphingolipids, cholesterol, and protein receptors which favor multiple cell signaling interactions at the cell membrane, necessary for signal transduction (1394, 1395). The exoplasmic leaflet is enriched with glycosphingolipids and sphingomyelin, the cytoplasmic leaflet is enriched with glycerolipids; cholesterol is present in both. The lipid composition makes this system highly dynamic in that several proteins act as structural proteins, transmembrane signaling proteins, and protein anchors linked to protein-protein interactions (1396).
Cholesterol in lipid rafts facilitates the clustering of α- and β-secretases. Although cholesterol does not have a direct effect on γ-secretases, the increment of local cholesterol facilitates the production of β-amyloid (1397, 1398).
The lipid composition of membranes is altered in sAD. β-amyloid oligomers and peptides are recruited in lipid rafts (1399-1402). Altered lipid raft composition also occurs in APP/PS1 double-transgenic mice, a model of familial cerebral β-amyloidopathy. Altered composition of lipid rafts is observed at the age of three months in parallel with the appearance of the first plaques in these mice (1403).
Prion protein expression in SPs further supports alteration of lipid raft-enriched cell membrane in association with β-amyloid deposition at advanced stages of sAD (128, 1404, 1405).
Lipid raft alteration in AD does not occur abruptly. The composition of lipid rafts changes with age in a gender-dependent manner. The main changes affect levels of plasmalogens, polyunsaturated fatty acids (especially DHA and AA), total polar lipids (mainly phosphatidylinositol, sphingomyelin, sulfatides, and cerebrosides), and total neutral lipids (particularly cholesterol and sterol esters) (1406).
Importantly, the lipid composition of lipid rafts in the entorhinal cortex and frontal cortex, but not the cerebellum, is already altered in human brain at NFT stages I-II without β-amyloid deposits; lipid rafts at these stages display higher anisotropy, indicating that lipid changes in brain at NFT stages I-II increase membrane order and viscosity in these domains (1407). Among other alterations, the structure of lipid rafts at stages I-II is associated with increased BACE1/AβPP interaction (1408).
There is also a close functional relationship between cytoskeletal proteins and cell membranes through protein-protein interactions, electrostatic interactions with lipid membranes, and lipid tails. These complementary interactions are ruined once one of the components is altered. Altered composition of lipid rafts and membrane proteins increases Aβ pathways and tau fibrilisation. At the same time, tau monomers and β-sheet-rich tau structures disrupt cell membranes (156, 212, 1389, 1409-1413). Tau connections with lipid tails depend on electrostatic interactions and phospholipid composition with high affinity for anionic lipids and anionic vesicles (1414, 1416). Tau monomers may concentrate at the membrane and form oligomers and fibrils under pathological conditions. These β-sheet-rich tau structures are capable of disrupting membrane organization and function (212). Deposition of abnormal tau at the pre- and post-synaptic membranes may appear prior to the appearance of NFTs, and it contributes to early synaptic dysfunction (409). Conversely, there is also the possibility that alterations at the cell membrane involving the lipid composition and post-translational modifications of membrane proteins trigger abnormal phosphorylation of tau and induce formation of tau fibrils (1417) (see section 21h).
Plasma membrane specializations containing caveolin are invaginated and form caveolae which are closely related to lipid rafts (1418). Caveolae in sAD participate in a wide number of processes including internalization of tau oligomers and β-amyloid metabolism (1419-1421).
Other factors influence membrane damage, in particular, microglial pro-inflammatory mediators generating membrane damages (1422).
22d. Mitochondria
Mitochondrial alterations were identified in the pioneering ultrastructural research of AD and were later sustained by functional studies (1423-1427). Mitochondrial alterations in sAD led to the formulation of the “mitochondrial cascade hypothesis”, proposing that mitochondrial alterations and failed energy metabolism trigger sAD (1428, 1429). Recent reviews have stressed the importance of mitochondrial failure in the pathogenesis of AD (1430-1432). Functional defects in ATP-synthase are considered a main contributory factor to explain failure of energy production in mitochondria (1433). Other studies have stressed the impairment of complex I in mild cases of AD (1434). Besides energy metabolism deterioration, abnormal mitochondrial function also causes increased production of ROS (1435-1437).
Mitochondrial ATP-synthase is a target of oxidative damage in the entorhinal cortex at stages I-II of NFT pathology; total levels of ATP-synthase are preserved but ATPase function is impaired (1438). Altered mitochondrial DNA methylation is manifested in the entorhinal cortex at stages I-II of NFT pathology and advances with disease progression (1028). Increased mitochondrial 5-methylcytosine levels are found in the D-loop region of mtDNA in the entorhinal cortex. Interestingly, this region shows a dynamic pattern in the content of mitochondrial 5-methylcytosine in APP/PS1 transgenic mice in parallel with the progression of β-amyloid pathology in these mice (1028). Aβ deposits have been considered a main cause of mitochondrial dysfunction in AD (1439). Additionally, mitochondrial abnormalities are observed in tau transgenic mice (1440). Aβ and tau pathology have synergistic effects on mitochondria in triple transgenic mice (1441). It has been proposed that there are mitochondrial links between brain aging and AD (1442).
The mitochondria-associated lipid raft-domain of the ER in close contact with the mitochondria, called MAM (mitochondria-associated ER membrane), facilitates functional and biochemical interaction between these structures, mainly linked to metabolism of the cholesterol, phospholipids, glucose, fatty acids, and calcium signaling (1443, 1444). MAM functions are altered at early stages of sAD (1445, 1446), thus prompting the “MAM-hypothesis” as a determinant in the pathogenesis of sAD (1447). There is growing evidence of impaired physical and proteome crosstalk between ER and mitochondria in sAD (1444).
22e. Oxidative stress damage
The mitochondria respiratory chain generates ROS which participate in cell signalling under physiological conditions. Peroxisomes, ER, microsomes, nucleus and plasma membrane are potential sources of ROS. Excess production of ROS and deficient anti-oxidant responses lead to oxidative stress damage to DNA, RNA, carbohydrates, lipids, and proteins. Ion-catalyzed oxidation of some amino acid residues may result in the production of carbonyl derivatives such as glutamic semialdehyde and aminoadipic semialdehyde. In addition to direct effects, oxidative modifications may induce the production of reactive carbonyl species such as glyoxal, glycoaldehyde, methylglyoxal, malondialdehyde (MDA), and 4-hydroxynonenal (HNE), derived from the oxidation of carbohydrates and lipids. Carbonyl species react with lysine, arginine, and cysteine residues, leading to the formation of advanced glycation and lipoxidation end-products (AGE/ALEs) in proteins. Typical AGEs/ALEs adducts are carboxymethyl-lysine (CML), carboxyethyl-lysine (CEL), and MDA-lysine (MDAL), among others (1357, 1373, 1381, 1448, 1449). Regarding RNS, nitric oxide damage to thiols, amines, and hydroxyls leads to nitrosative damage. Reactions with RNS lead to the formation of 3-nitrotyrosine (nitration) and to oxidation of distinct substrates. Reactive peroxinitrite is able to nitrate tyrosine residues and to oxidize methionine residues of proteins (1450).
Oxidative stress damage is a major component of brain aging. Protein oxidative and glycoxidative damage significantly increases with age; 60 years of age is the breakpoint of human frontal cortex aging (1451).
Oxidative stress damage in the aging brain is region- and age-dependent (1452). Regional vulnerability to neurodegeneration based on energy demands, oxidative stress, and other metabolic factors can predict neurodegeneration (1453). For example, lower energy demand, lower mitochondrial stress, and one-carbon metabolism (particularly restricted to the methionine cycle), together with lower target of rapamycin (TOR) signaling and better antioxidant capacity, occur in the frontal cortex compared with the entorhinal cortex and the hippocampus. These differences suggest that the frontal cortex is relatively resistant to stress compared to the entorhinal cortex and hippocampus (1454).
However, this hypothesis does not apply universally when assessing different brain regions of individual brains in parallel (1455). The results are more complex and their interpretation is more difficult. Vulnerable cortical and diencephalic regions are in fact more resistant to degeneration in aging (1455). However, differences in vulnerability to protein oxidation are dependent on the subcellular localization, secondary structure, and external exposition of certain amino acids. Lipoxidized proteins are mainly those involved in energy metabolism, cytoskeleton, proteostasis, neurotransmission, and O2/CO2/heme metabolism (1452).
AGEs are produced in aging and sAD, but their levels and the levels of their receptor (RAGE) do not correlate with Aβ levels, tau levels, or dementia (1456, 1457).
Most studies of oxidative stress damage in sAD are centered on stages V-VI (cases with dementia) and III-IV (cases with MCI). Proteins involved in glycolysis and energy metabolism, in electron transport chain, oxidative phosphorylation, and other mitochondrial components; structural proteins; chaperones; stress proteins, and stress responses; and proteins of the UPS are all targets of oxidative damage (1373, 1458-1467). Protein oxidative damage is usually accompanied by decreased functional activity (1468). Altered enzymatic activity has been demonstrated for the oxidized protein creatine kinase BB, enolase 1, glutamine synthetase, Pin-1, carbonic anhydrase 2, UCHL-1, α-enolase, GAPDH, GDH, H+ transporting ATPase, LDH, ATP synthase, and pyruvate kinase in sAD. A few studies have included the identification of oxidatively damaged protein, the quantification of total protein levels, and the reduction of enzymatic activity (1381). In an attempt to correlate oxidative stress damage and regional vulnerability in sAD, a meta-analysis of MDA, HNE, protein carbonylation, 8-hydroxyguanine levels and superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase activities showed that changes linked to oxidative stress were variable from one region to another and dependent on the type of adduct. No correlation was seen between oxidative damage and regional vulnerability (1469).
Oxidative damage was advocated to be the earliest event in sAD (1470). Further research revealed that oxidative damage was more marked in younger cases, in cases with rapid disease progression, and in neurons without NFTs when compared with neurons with tangles in the same individual (1471). Moreover, oxidative stress precedes β-amyloid deposition in pre-symptomatic fAD and Down syndrome (1472, 1473). Early observations suggested that oxidative post-translational modifications might play a role in the formation of SPs and NFTs (1474). Following this argument, aggregation of Aβ and tau was considered a compensatory response to underlying oxidative stress (1475). However, β-amyloid is also a cause of oxidative stress, thereby potentiating the loop (1476). Oxidative stress generates mitochondrial dysfunction by damaging structural proteins and components of the mitochondrial respiratory chain in sAD (1438, 1477-1479). Oxidative damage also causes synaptic dysfunction (425).
As indicated in previous paragraphs, oxidative damage of ATP-synthase resulting in the loss of its function occurs at stages I-II of NFT pathology (1438). Mitochondrial dysfunction, abnormalities in lipid rafts, and oxidative stress damage potentiate each other and are major players in neuronal energy failure at the first stages of NFT pathology (1480). Another relevant consequence of oxidative stress is the effect of advanced glycation end products on cell-cycle re-entry and arrest, also occurring at the first stages of sAD (1481).
22f. Inflammation
Aging is accompanied by low levels of activated innate inflammatory responses (1482, 1483).
The role of microglia and inflammation in the aging brain and sAD has been discussed in previous paragraphs. Here, the focus is on the relevance of inflammatory changes in brain aging and at early stages of AD pathology. Several reports in the 1990s described a protective effect of non-steroidal anti-inflammatory drugs used for the treatment of autoimmnune diseases on the manifestation and progression of sAD (1484-1490). Further studies delineated their positive effects when the treatment was initiated before the appearance of cognitive impairment, but the effects of the same treatments were minimal when administered in advanced AD. These observations show that inflammatory responses modulate the history of AD at the early stages of the process. More importantly, there is also a chance for anti-inflammatory drugs in the treatment of sAD when administered at the appropriate time (1491-1493).
Molecular studies disclosed that multiple cytokines are involved at the early/middle stages of AD (1494). More detailed studies in MA and in cases at first (I-II), middle (III-IV), and advanced (V-VI) NFT stages examined the expression of cytokines and mediators of the immune system in different regions progressively affected in sAD: the entorhinal cortex, orbitofrontal cortex, and frontal cortex area 8 (838). Changes in mRNA expression correlated with the corresponding protein levels as revealed by immunohistochemistry and western blotting (838). Moreover, gene regulation at first stages of AD pathology (NFT I-II, SPs: 0) was not related to NFTs, β-amyloid plaques, concentration of Aβ40 and Aβ42, or membrane-bound fibrillar β-amyloid in the frontal cortex (838). These observations do not contradict previous studies showing a relationship between membrane-associated β-amyloid and inflammatory changes in cases with more advanced preclinical AD and SPs in the cerebral cortex (101), but they highlight the observations that: (a) inflammatory markers appear at the first stages of NFT pathology (stages I-III) in regions with no NFTs and SPs; (b) inflammatory changes are modified with disease progression; and (c) different inflammatory responses occur simultaneously in different regions in the same individual.
22g. Protein synthesis impairment
Protein synthesis is altered in sAD, and this is due to multiple alterations at different subcellular levels from the nucleolus to the ribosome. The relation nuclear organizer region (NOR) surface/total nucleus surface is reduced; the rDNA promoter is hyper-methylated, and dimethylated histone H3K9 and acetylated histone H3K12 are decreased in the CA1 region of the hippocampus; nuclear tau declines; specific transcription factors are abnormally regulated; rRNA levels are decreased and RNA is oxidatively damaged; the expression of nucleolar proteins as well as the expression of RNAs involved in the generation of ribosomal proteins decreases; the expression levels of translation initiation and elongation factor of the protein synthesis in ribosomes is dysregulated; and the capacity of isolated ribosomes to incorporate S35 methionine into protein is impaired (1010, 1473, 1495-1506). Tau protein disrupts nucleocytoplasmic transport in AD (1507), but the role of tau, if any, in the other steps of the protein synthesis pathway is not known.
Alterations of protein synthesis pathways are already identified in the hippocampus at the first stages of NFT pathology. Nucleophosmin 1 (NPM1) mRNA is significantly increased, and upstream binding transcription factor RNA polymerase I gene (UBTF) mRNA and 28S rRNA significantly decreased in CA1, but not in the dentate gyrus at NFT stages I-II. Dimethylated histone H3K9 (H3K9m2) immunoreactivity is reduced in neurons of the dentate gyrus and CA1 at NFT stages I-II. mRNA expression of ribosomal proteins RPL23A, RPL26, RPL31RPS5, RPS6, RPS10, and RPS13 is significantly reduced in the dentate gyrus at stages I-II when compared with MA individuals without NFT pathology. In contrast, RPL5 and RPL26 mRNA expression is increased, and RPS5 and RPS6 mRNA decreased in CA1 at the same stages I-II (1508). These results show early alterations not only in the CA1 region, which will be involved in NFT pathology, but also in the dentate gyrus, which does not contain abnormal tau deposits at any time in the process. Seven upregulated miRNAs (miR-125b, miR-146a, miR-200c, miR-26b, miR-30e, miR-34a, and miR-34c) and three downregulated miRNAs (miR-107, miR-210, and miR-485), all of which associated with oxidative stress, are found in vulnerable brain regions of sAD at the clinical prodromal stage (1501). Together, these observations show that alterations in protein synthesis pathways appear at early stages of AD-related pathology and they are not linked to tau and β-amyloid deposits.
Disruption at many steps of the protein synthesis pathway increases in all regions with sAD progression (1508), making it difficult to establish a causative relationship between tau and β-amyloid pathology and the progressive decay of protein synthesis at advanced stages of sAD.
22h. Dysregulated protein phosphorylation
Early phosphoproteomics studies identified a few abnormally phosphorylated proteins in the hippocampus and cerebral cortex in small numbers of cases with sAD compared with controls (1509-1511). Subsequent work with more accurate methods in advanced sAD identified a large number of abnormally phosphorylated proteins, some of them with increased phosphorylation, and others with decreased phosphorylation. Abnormally phosphorylated proteins corresponded to cytoskeletal proteins, integral membrane proteins, synaptic proteins, adhesion molecules, serine/threonine kinases, transport/cargo proteins, heat-shock proteins, and others mostly involved in cell growth and/or maintenance, cell communication, and metabolism (1417, 1513, 1514). Multi-omics integration highlighted relevant altered networks including amyloid cascade, inflammation, complement, WNT protein signaling, transforming growth factor-β and bone morphogenic protein signaling, lipid metabolism, iron homeostasis, and membrane transport (1515).
Recent studies performed in the entorhinal cortex and frontal cortex in human brain aging and sAD at different NFT stages identified sixty-five dysregulated phosphoproteins in the entorhinal cortex, and eighty-one phosphoproteins in the frontal cortex at NFT stages I-II when compared with MA individuals without NFT pathology. Dysregulated protein phosphorylation of selected proteins occurs in parallel with the appearance of NFTs in the entorhinal cortex but precedes the appearance of NFTs and SPs in the frontal cortex. The number of dysregulated phosphoproteins increases in both regions with NFT stage, most of them added to those already dysregulated at stages I-II. Considering the total number of identified dysregulated phosphoproteins, the most active period corresponds to NFT stages III-IV, at a time when a subpopulation of people might be clinically categorized as suffering from MCI (1417).
The main group of dysregulated phosphoproteins at NFT stages I-II are membrane proteins; proteins of the cytoskeleton; proteins of the synapses and dense core vesicles; proteins linked to membrane transport and ion channels; kinases; proteins linked to DNA and protein deacetylation; proteins linked to gene transcription and protein synthesis, and proteins involved in energy metabolism (1417). Altered phosphorylation of selected proteins, accomplished by activation of several kinases, may alter membrane and cytoskeletal function, among these synaptic transmission and membrane/cytoskeleton signaling, in addition to energy metabolism, protein synthesis, and DNA homeostasis (1417).
DAGs are constituents of cell membranes that participate in intermediate lipid metabolism, and they are key components in lipid-mediated signaling. In neurons, DAGs modulate several signal transduction proteins linked to the activation of protein kinases, traffic and fusion of synaptic vesicles, ion channels, axonal guidance, and cytoskeletal homeostasis, among others (1516-1518). Tau and β-amyloid phosphorylation may also be mediated by DAGs and PKC (1519). DAG levels are increased in the sAD frontal cortex (1520, 1521). Further studies are needed to learn about possible links between DAGs and abnormal phosphorylation of cell membrane proteins.
23. Concluding comments
Linear logic based on the assumption that a cause results in one or several effects does not explain complex biological phenomena such as brain aging and sAD; there is no single cause of aging and sAD. Rather, the biological processes involved in generation, development, living, decline, and death are complex concatenations of complementary, disruptive, and adaptive responses. Not surprisingly, the mutually exclusive hypotheses formulated to explain sAD are not satisfactory.
Genetic studies in sAD have also provided a wealth of information identifying genetic risk factors which principally cover cholesterol and lipid metabolism, inflammation, and cell membranes and synapses (1390).
A scheme of the several factors involved in brain aging and sAD is presented in figure 9. In addition to genetic factors, there is an interaction between molecular alterations in cellular structures that may favor the production of β-amyloid and abnormal tau. Conversely, the presence of β-amyloid and abnormal tau has a negative effect on the majority of cellular structures, contributing to a detrimental loop in sAD pathogenesis.
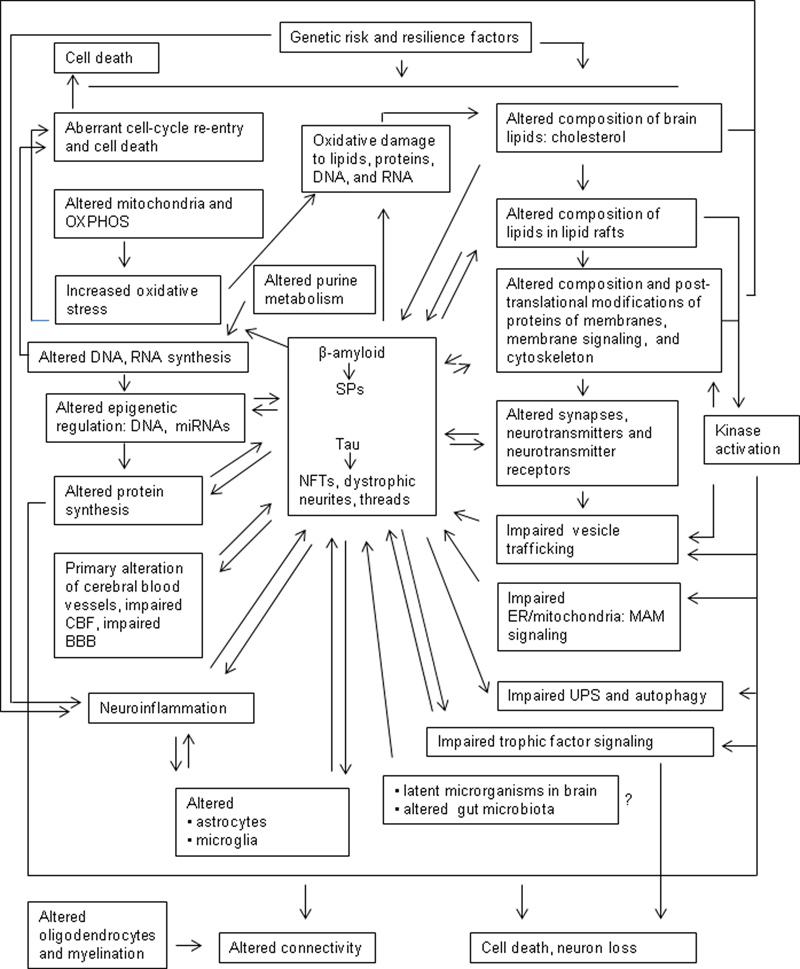
Figure 9: Factors involved in brain aging and sAD. In addition to genetic factors, molecular alterations in cellular structures may induce the production of β-amyloid and abnormal tau. Conversely, the presence of β-amyloid and abnormal tau has a negative effect on the majority of cellular structures, contributing to a harmful loop in sAD pathogenesis.
Molecular alterations are similar and cumulative in brain aging and sAD. Therefore, a continuum of brain aging and sAD primarily based on an archetypal distribution of NFTs usually followed by the deposition of β-amyloid plaques is a factual possibility.
The pathogenesis and evolution of arteriosclerosis is another example of cumulative age-related degenerative changes in blood vessels. Almost all people are affected to some degree by the age of 65. However, the first lesions in atherosclerosis, characterized by macrophage infiltration and intracellular lipid accumulation in the blood vessel wall, may occur in the first or second decade; inflammation, endothelial and perycite damage, altered metabolism of muscle fibers, extracellular lipid cores and atheromatous plaques develop later. Clinical manifestations may appear at advanced stages of fibroatheroma, as well as complicated lesions in MA individuals. Thus, the atheromatous plaque, the characteristic lesion of atherosclerosis, is not the first requirement to identify atherosclerosis as a biological process which is expressed by cumulative stages of blood vessel damage leading or not to clinical manifestations of variable severity.
Searching possible therapies for sAD has been constant over the years. Cholinesterase inhibitors, NMDA receptor antagonists, membrane protectors, anti-oxidants, and anti-inflammatory agents, all assayed at middle and advanced clinical stages of sAD, have proven poorly effective. Other treatments are directed to reducing β-amyloid or tau accumulation. Several Aβ-protein-targeted drugs, including β-secretase inhibitors, γ-secretase inhibitors and modulators, α-secretase activators, direct inhibitors of Aβ aggregation, and immunotherapy have been assayed or are under different phases of clinical trial (1522). β-amyloid immunization in humans has been successful in the sense that SPs are largely reduced in the brain of treated patients; yet the abundance of tau deposits and the progression of the disease are not substantially modified by β-amyloid immunization (719, 1523, 1524). The clinical improvement seen with β-amyloid immunotherapy has been null or negligible, or at best arguable (1525-1530). Treatment with a BACE1 inhibitor has yielded very limited neuropathological improvement (1531). Trials with new generation Aβ immunotherapy are in progress.
Following the same line of thinking, tau immunization, although putatively preventing tau accumulation, is unlikely to be a unique alternative treatment (1532-1534). Various tau-based therapies have been developed or are under development. These therapies include the use of inhibitors of tau phosphorylation, glycosylation, and acetylation; microtubule stabilizers; inhibitors of tau aggregation; and anti-tau immunotherapy (1535-1538). Various tau therapies based on active and passive immunization are effective in murine and primate models. However, some of these attempts have failed in sAD (1539). Although encouraging, it is not clear how such treatments will reduce the different forms of abnormal tau and decrease Aβ burden. In short, there is no evidence that merely lessening the abnormal levels and deposits of tau or β-amyloid at middle and advanced stages of the illness will cure sAD. Therefore, it is crucial to re-consider the optimal age to start combined anti-tau- and anti-β-amyloid-based treatments to combat two main representative components of AD. The identification of new putative targets for therapeutic intervention before the appearance of tau and amyloid deposits is a promising endeavor.
Gene therapy has also been assessed in transgenic mouse models with variable success using both vector-based therapies and genetically modified cell replacement (1540-1542). Studies in humans are limited, at this time, to upregulated NGF in sAD patients, with little evidence of benefit (1543-1545).
Exosomes are a class of membrane vesicles derived from endolysosomal compartment implicated in cell-cell communication by shuttling different lipids, protein, and RNAs between cells. The use of shuttles including exosomes to deliver selected molecules into the cells is still at the early stages.
The meager success of available therapies to curb the effects of human brain aging and sAD is a matter of concern. Besides disease modification and symptomatic therapies for sAD (1546-1549), other relevant and long-term ventures may also be considered.
One of these is a revision of animal models used to test sAD therapies, because they have failed when applied to humans (1550). To make this clear: for 25 years, many AD-like mouse models have been “cured” in the lab, only for the treatments to fail in clinical trials in humans (1551, 1552). It seems that mice have the ability to remodel complex metabolic defects once one of the factors involved in the process is normalized. This does not occur in humans.
These considerations are made yet more compulsory because a number of molecular function derangements take place before or concurrent with the appearance of β-amyloid and tau pathology morphologically manifested as NFTs and SPs. Altered lipid and protein composition of cellular membranes accompanied by impaired subcellular cell signaling from the cell membrane to the ER, mitochondrial membranes, altered synapses, altered mitochondria and energy metabolism, and reactivation and abortion of developmental programs leading to neuronal death, together with dysfunctional BBB and singular brain inflammatory expression, all point to sAD as a human brain age-related disorder of convergent mechanisms that shatters brain self-organization (329, 1343, 1553, 1554).
A great data analysis covering the tremendous body of information on AD must be updated in real time.
We need to think of the coming decades as an opportunity to take advantage of the rapid growth of artificial intelligence (AI) and cell reprogramming. We will be able to redesign some aspects of the human brain in the near future using advanced technologies. To this end, we also need to identify the main targets and appropriate intervention times. Timing is crucial since it will be difficult to reprogram molecular pathways in old-adult individuals that have already suffered brain deterioration. Brain reprogramming would likely be undertaken before the beginning of the slow-pace functional decline in MA adults. Brain reprogramming may cover different areas including brain DNA editing, utilization of external electrical or wave-based signals to reduce energy consumption of basic neuronal networks, optimization of mitochondrial function, implanting of microdevices to facilitate cooperative human-machine function, pharmacological combined protection of lipid-protein interactions, and program resetting during stages of sleep. Improvement of brain function in aging and sAD has a chance in the application of high-throughput molecular technology, AI, and robotics (1394, 1555-1561). This new era is certain to present formidable ethical challenges.
Abbbreviations
2-AG - arachidonoyl glycerol, 5-HT - 5-hydroxytriptamine, 3’-UTR - complementary un-translated regions, AA - arachidonic acid, Aβ - beta amyloid, ABCA7 - ATP binding cassette subfamily A member 7, AβOs - soluble β-amyloid oligomers, AC - adenylyl cyclise, ACh - acetylcholine, AChE - acetylcholinesterase, AD - Alzheimer disease, ADAM10 - ADAM metallopeptidase domain 10, ADDLs - amyloid-β derived diffusible ligands, ADNC - AD-neuropathologic change, AEA - N-arachidonoyl ethanolamine, AGD - argyrophylic grain disease, AGEs - advanced glycation end-products, Ago - argonaute subfamily proteins, AI - artificial intelligence, ALEs - advanced lipoxidation end-products, AMP - adenosine monophosphate, AMPA - α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, AMPAR - α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, APHH1A - α-secretase subunit A, ApoE - apolipoprotein E, APP - amyloid precursor protein, AQP - aquaporin, ARTAG - aging-related tau astrogliopathy, ASC - inflammasome-adaptor protein, ATF-6 - activating transcription factor 6, ATP8B1 - ATPase phospholipid transporting 8B1, ATP - adenosine triphosphate, BACE - β-secretase, β-AR - β-adrenergic receptor, BBB - blood brain barrier, BDNF - brain-derived neurotrophic factor, BIN1 - bridging integrator 1, C1QTNF7 - complement C1q tumor necrosis factor-related protein 7, C3AR1 - complement C3a receptor 1, Ca2+ - calcium ion, cAMP - cyclic adenosine monophosphate, CB - calbindin D28K, CBF - cerebral blood flow, CBR - cannabinoid receptor, CD2AP - CD2-associated protein, CDK5 - cyclin-dependent kinase 5, CERAD - Consortium to Establish a Registry for Alzheimer’s Disease, CEL - carboxyethyl-lysine, cGMP - cyclic guanosine monophosphate, ChAT - choline acetyl transferase, CK1-δ - casein kinase I isoform delta, CLU - clusterin, CML - carboxymethyl-lysine, CMV - human cytomegalovirus, CNP - 2',3'-cyclic-nucleotide 3'-phosphodiesterase, CNS - central nervous system, COMT - catechol-O-methyltransferase, CpG - cytosine-guanine-rich regions , CR - calretinin, CR1 - complement component (3b/4b) receptor 1, CREB - cAMP response element-binding protein, CSF - cerebrospinal fluid, CSF1R - colony stimulating factor 1 receptor, CSF3R - colony stimulating factor 3 receptor, CT - computed tomography, DAG - diacylglycerol, DBH - dopamine β-hydroxylase, DCX - doublecortin, DHA - docosahexanoic acid, DNMT - DNA methyl transferases, GSH - glutathione, EAAT - excitatory amino acid transporter, EBV - Epstein-Barr virus, EOFAD - early onset Alzheimer disease, Eph - ephrin, EPHA1 - ephrin receptor A1, ER - endoplasmic reticulum, exRNAs - extracellular RNAs, fAD - familial Alzheimer disease, FAD - flavin adenine dinucleotide, FBD - familial British dementia, FcγRIIb - FC&gamma, receptor II-b, FDD - familial Danish dementia, GABA - γ- aminobutyric acid, GABAA receptors - ionotropic GABAA receptors, GABAB receptors - metabotropic GABAB receptors, GAD - glutamic acid decarboxylase, GAP-43 - growth-associated protein 43, GDH - glutamate dehydrogenase, GFAP - glial fibrillary acidic protein, GluRs - glutamate receptors, GLUT 1 - glucose transporter 1, GMP - guanosine monophosphate, GPCR - G-protein-coupled receptor, GRP78/BiP - glucose-related protein 78, GSK3β - glycogen synthase kinase 3β, GSS - Gerstmann-Sträussler-Scheinker, GVD - granulovacuolar degeneration, GWAS - genome-wide association studies, H3K4me1 - methylation of lysine 4 of histone 3, H3K4m3 - trimethylation of histone 3 at lysine 4, H3K9me2 - dimethylation of histone H3 at lysine 9, HAT - histone acetyltransferase, HDAC - histone deacetylase, HDM - histone demethylase, HHV-6 - human herpes virus 6, HMT - histone methyltransferase, HNE - 4-hydroxynonenal, HSV-1 - herpes simplex virus 1, iCJD - iatrogenic Creutzfeldt-Jabob disease, IDE - insulin degrading enzyme, IFN-γ - interferon γ, iGluR - ioinotropic glutamate receptor, IL - interleukin, iPSC - indiuced pluripotent stem cells, IRE 1 - inositol-requiring protein 1, Jak2 - janus kinase 2, KA - kainate, KAR - kainate receptor, LAMP-1 - lysosomal-associated membrane protein 1, LC3 - protein light chain 3, LC-MS/MS - liquid chromatography (LC) tandem mass spectrometry (MS), lcnRNAs - long non-coding RNAs, LDH - lactate dehydrogenase, LDLR - low density lipoprotein protein receptor 1, LilrB2 - PirB human orthologue receptor , LOAD - late onset Alzheimer disease, LP2 - lipoprotein lipase 2, LRP1 - HLDL receptor related protein 1, MA - middle-aged, mAChR - muscarinic acetylcholine receptor, MAG - myelin associated glycoprotein, MAL - myelkin and lymphocyte protein, MAMs - mitochondria-associated ER membranes, MAP2 - microtubule associated protein 2, MAPK - mitogen-activated protein kinase, MAPT - microtubule associated protein tau, MAPT H1 - microtubule-associated protein tau haplotype 1, MAO - mono-amino oxidase, MBP - myelin basic protein, MCI - mild cognitive impairment, MDA - malondialdehyde, MDAL - MDA-lysine, MEF2C - myocyte-specific enhancer factor 2C, Mg2+ - magnesium ion, mGluR - metabotropic glutamate receptor, miRNA - microRNA, MLKL - mixed lineage kinase domain-like protein, MOBP - myelin-associated oligodendrocyte basic protein, MOG - myelin-oligodendrocyte glycoprotein, MRI - magnetic resonance imaging, MSA4 - membrane-spanning 4-domains subfamily A, mtRNAs - mitochondrial RNAs, MUFA - monounsaturated fatty acids, nAChR - nicotinic acetylcholine receptor, NADþ - nicotinamide adenine dinucleotide, NADPþ - nicotinamide adenine dinucleotide phosphate, NCT/NCSTN - nicastrin, NEP/NME - neprelysin, NFT - neurofibrillary tangle, NG2 - neural/glial antigen 2, NGF - nerve growth factor, NIA-AA - National Institute on Aging–Alzheimer’s Association, NLRP3R - pyrin-domain containing 3, NMDA - N-methyl D-aspartate, NMDAR - N-methyl-D-aspartate receptor, NOR - nuclear iorganizer region, NPM1 - Nucleophosmin 1, nRNAs - small non-coding RNAs, OPC - oligodendroglial precurosor cells, PART - primary age-related tauopathy, P75NTR - p75 neurotrophin receptor, low affinity nerve growth factor receptor, PDGFRA - platelet-derived growth factor receptor A, PEN2/PSENEN - presenilin enhancer γ-secretase subunit, PERK - PKR-like endoplasmic reticulum kinase, PET - positron emission tomography, PHF - paired helical filament, PI3K - phosphatidylinositol 3-kinase, PI3P - phosphatidylinositol trisphosphate, PICALM - phosphatidylinositol binding clathrin assembly protein, PiD - Pick disease, PILRA - paired immunoglobin like type 2 receptor alpha, PirB - paired immunoglobulin-like receptor B, piRNAs - PIWI-interacting RNAs, PKA - protein kinase A, PKC - protein kinase C, PLC - phospholipase C, PLP1 - proteolipid protein 1, PPAR - peroxisome proliferator-activated receptors, PRNP - prion protein gene, PrPC - cellular prion protein, PSEN1 - presenilin 1, PSEN2 - presenilin 2, PTK2B - protein tyrosine kinase 2 beta, PUFA - polyunsaturated fatty acids, PV - parvalbumin, RAB10 - Ras-related protein Rab-10, RAGE - AGE receptor, REST - RE1-silencing transcription factor, RIP3 - receptor interacting protein 3, RNS - reactive nitrogen species, RORB - RAR related orphan receptor B, ROS - reactive oxygen species, RP - ribosomal protein, rpAD - rapidly progressive AD, rRNA - ribosomal RNA, sAD - sporadic Alzheimer disease, SAM - S-adenosyl-L-methionine, SAPK/JNK - stress-activated protein kinase/JUN N-terminal kinase, scaRNAs - small Cajal-body specific RNAs, SFPQ - splicing factor proline and glutamine rich, siRNAs - small interfering RNAs, SNAP - soluble NSF attachment protein receptor, SNARE - SNAP receptor protein, snRNAseq - single nuclei RNA sequencing, snRNA - small nuclear RNAs, snoRNAs - small nucleolar RNAs, SORL1 - sortilin-related receptor 1, SPI1 - Spi-1 proto-oncogene, SP - senile plaque, STAT1 - signal transducer and activator of transcription 1, TET1 - ten-eleven-translocation-1 protein, TGF - transforming growth factor, TLR - Toll like receptor, TNF - tumor necrosis factor, TOR - target of rapamycin, TRAF2 - TNF receptor-associated factor 2, TREM2 - triggering receptor expressed on myeloid cells 2, TrkA - specific NGF receptor, tRNA - transfer RNAs, UBTF - upstream binding transcription factor RNA polymerase I gene, UCHL-1 - ubiquitin C-terminal hydrolase L1, UPR - misfolded protein response, UPS - ubiquitin proteasome system, VGAT - vesicular GABA transporter, VGLUT - vesicular glutamate transporter, VMAT - vesicular monoamino transporter, WM - white matter, WT - wild type, XBP-1 - X-box binding protein 1.
Funding
The project leading to these results received funding from the “la Caixa” Foundation (ID 100010434) under the agreement LCF/PR/HR19/52160007, HR18-00452. I thank the CERCA programme of the Generalitat de Catalunya for institutional support.
Acknowledgements
This work is, in part, the result of the personal experience of 40 years in the study of AD and other tauopathies. To name all the people I would like to thank for their help, support, and satisfactory collaboration is practically impossible. I appreciate the aid of CIBERNED, and I am extremely grateful to Tom Yohannan for his continual editorial assistance.
References
- Duyckaerts C, Dickson DW. Neuropathology of Alzheimer’s disease and its variants. In: Neurodegeneration: the molecular pathology of dementia and and movement disorders; second edition. Dickson DW, Wetter RO (eds) International Society of Neuropathology, Blackwell Publish Ltd, 2011, pp: 62-91. https://doi.org/10.1002/9781444341256.ch10
- Knopman D. Clinical aspects of Alzheimer’s disease. In: Neurodegeneration, the molecular pathology of dementia and movement disorders. Dickson DW, Weller RO (eds) Wiley- Blackwell, 2011, pp: 39-50. https://doi.org/10.1038/s41572-021-00269-y
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1:a006189. https://doi.org/10.1101/cshperspect.a006189
- Love J, Kalaria R. Dementia. In: Greenfield’s neuropathology, vol 1, nnth edition. Love S, Budka H, Ironside JW, Perry A (eds). CRC Press, 2015, pp: 858-973.
- Lowe J, Kalaria R. Dementia. In: Greenfield’s neuropathology. Ninth edition. Loe S, Budka H, Ironside JW, Perry A (eds). CRC Press 2015, pp: 858-973.
- Masters CL, Bateman R, Blennow K, et al. Alzheimer's disease. Nat Rev Dis Primers 2015;1:15056. https://doi.org/10.1038/nrdp.2015.56
- DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodeg 2019;14:32. https://doi.org/10.1186/s13024-019-0333-5
- Jellinger KA. Neuropathology of the Alzheimer’s continuum: an update. Free Neuropathol 2020;1:32. https://doi.org/10.17879/freeneuropathology-2020-3050
- Avila J, Perry G. A multilevel view of the development of Alzheimer’s disease. Neuroscience 2021; 457: 283-293. https://doi.org/10.1016/j.neuroscience.2020.11.015
- Blocq P, Marinesco G. Sur les lésions et la pathogénie de l’épilepsie dite essentielle. Sem Méd 1892; 12:445-446.
- Redlich E. Uber miliare Sklerose der Hirnrinde bei seniler Atrophie. Jahrb Psychiat Neurol 1898;17:208-216.
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiat 1907; 64: 146-148.
- Perusini G. Über klinisch und histologisch eigenartige psychische Erkrankungen des spaateren Lebensalters. In: Nissl F, Alzheimer A (eds) Histologische und histopathologische Arbeiten. Fischer, Jena, 1910
- Kraepelin E. Psychiatrie: ein Lehrbuch für Studierende und Ärzte. II. Band, Klinische Psychiatrie. Verlag Johann Ambrosius Barth, Leipzig, 1910, vol.II.
- Fischer O. Miliare Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Monatsschr Psychiat Neurol 1907; 22: 361-372. https://doi.org/10.1159/000211873
- Fischer O. Die presbyophrene Demenz, deren anatomische Grundlage und klinische Abgrenzung. Z ges Neurol Psychiat 1910;3: 371-471. https://doi.org/10.1007/BF02893605
- Fischer O. Ein weiterer Beitrag zur Klinik und Pathologie der presbyophrenen Demenz. Z ges Neurol Psychiat 1912; 12: 99-135. https://doi.org/10.1007/BF02866372
- Simchowitz T. Histologische Studien uber die senile demenz. In: Nissl F, Alzheimer A (eds) Histologische und Histopathologische Arbeiten über die Grosshirnrindem, Vol. 4. Fischer, Jena, 1911; pp 267-444.
- Goedert M. Oskar Fischer and the study of dementia. Brain 2009; 132;1102-1111. https://doi.org/10.1093/brain/awn256
- Bielschowsky M. Zur Kenntnis der Alzheimerschen Krankheit (präsenilen Demenz mit Herdsymptomen). J Psychol Neurol 1911;18:1-20.
- Karenberg A. Early history of Pick's disease. Fortschr Neurol Psychiatr 2001;69: 545-50. https://doi.org/10.1055/s-2001-18378
- Delay J, Brion S. Les démences tardives. Masson, Paris, 1962
- Jervis GA, Soltz SE. Alzheimer’s disease: The so-called juvenile form. Am J Psychiatr 1936; 93: 39-56. https://doi.org/10.1176/ajp.93.1.39
- Blessed G, Tomlinson B, Roth M. The association between quantitative measures of dementia and of senile changes in the cerebral grey matter of elderly subjects. Br J Psych 1968;114: 797-811. https://doi.org/10.1192/bjp.114.512.797
- Katzman R. Editorial: the prevalence and malignancy of Alzheimer disease: a major killer. Arch Neurol 1976; 33:217-218. https://doi.org/10.1001/archneur.1976.00500040001001
- Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci 1970; 11:205-242. https://doi.org/10.1016/0022-510x(70)90063-8
- Terry RD, Katzman R. Senile dementia of the Alzheimer type. Ann Neurol 1983;14: 497-506. https://doi.org/10.1002/ana.410140502
- McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263-269. https://doi.org/10.1016/j.jalz.2011.03.005
- Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease Neurology 1988;38:1682-1687. https://doi.org/10.1212/wnl.38.11.1682
- Price JL, Davis PB, Morris JC, et al. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging 1991;12:295-312. https://doi.org/10.1016/0197-4580(91)90006-6
- rriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology 1992;42:1681-1688. https://doi.org/10.1212/wnl.42.9.1681
- Bouras C, Hof PR, Morrison JH. Neurofibrillary tangle densities in the hippocampal formation in a non-demented population define subgroups of patients with differential early pathologic changes. Neurosci Lett 1993;153:131-135. https://doi.org/10.1016/0304-3940(93)90305-5
- Bouras C, Hof PR, Giannakopoulos P, et al. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex 1994;4:138-150. https://doi.org/10.1093/cercor/4.2.138
- Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087-1095. https://doi.org/10.1093/jnen/62.11.1087
- Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837-1844. https://doi.org/10.1212/01.wnl.0000219668.47116.e6
- Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol 1985;42: 1097–1105. https://doi.org/10.1001/archneur.1985.04060100083029
- Wisniewski HM, Robe A, ZigmanW, et al. Neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol 1989;48: 606-609. https://doi.org/10.1097/00005072-198911000-00001
- Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991;41:479-486. https://doi.org/10.1212/wnl.41.4.479
- Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer's disease. A primer for practicing pathologists. Arch Pathol Lab Med 1993;117:132-144. PMID: 8427562
- Hubbard BM, Fenton GW, Anderson JM. A quantitative histological study of early clinical and preclinical Alzheimer’s disease. Neuropathol Appl Neurobiol 1990;16:111–121. https://doi.org/10.1111/j.1365-2990.1990.tb00940.x
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997; 18:351-357. https://doi.org/10.1016/s0197-4580(97)00056-0
- Hulette C, Welsh-Bohmer K, Murray M, et al. Neuropathological and neuropsychological changes in "normal" aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol 1998;57:1168-1274. https://doi.org/10.1097/00005072-199812000-00009 PMID: 9862640.
- Price J, Morris JC. Tangles and plaques in non demented aging and "preclinical" Alzheimer's disease. Ann Neurol 1999;45:358-368. https://doi.org/10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x
- Khachaturian ZS. Diagnosis of Alzheimer's disease: two-decades of progress. J Alzheimers Dis 2006;9:409-415. https://doi.org/10.3233/jad-2006-9s346
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol 2010;9:1118-1127. https://doi.org/10.1016/S1474-4422(10)70223-4
- Albert MS, Dekosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:270–279. https://doi.org/10.1016/j.jalz.2011.03.008
- McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263-269. https://doi.org/10.1016/j.jalz.2011.03.005
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280-292. https://doi.org/10.1016/j.jalz.2011.03.003
- Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1-13. https://doi.org/10.1016/j.jalz.2011.10.007
- Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1-11. https://doi.org/10.1007/s00401-011-0910-3
- Jack CR, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535-562. https://doi.org/10.1016/j.jalz.2018.02.018
- Ward A, Tardiff S, Dye C, et al. Rate of conversion from prodromal Alzheimer’s disease to Alzheimer’s dementia: A systematic review of the literature. Dement Geriatr Cogn Disord Extra 2013;3:320-332. https://doi.org/10.1159/000354370
- Petersen RC, Lopez O, Armstrong MJ, et al. Practice guideline update summary: Mild cognitive impairment. Neurology 2018;90:126-135. https://doi.org/10.1212/WNL.0000000000004826
- Gao S, Hendrie HC, Hall KS, et al. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psych 1998; 55:809-815. https://doi.org/10.1001/archpsyc.55.9.809
- Vermunt L, Sikkes SAM, van den Hout A, et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement 2019;15:888-898. https://doi.org/10.1016/j.jalz.2019.04.001
- Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 1963; 197: 192-193. PMID: 14032480.
- Kidd M. Alzheimer’s disease- an alectron microscopical study. Brain 1964;87:307-320. https://doi.org/10.1093/brain/87.2.307
- Terry RD. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol 1963;22:629-642. https://doi.org/10.1097/00005072-196310000-00005
- Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol 1964;44: 269-297. PMID: 14119171.
- Luse SA, Smith KR. The ultrastructure of senile plaques. Am J Pathol 1964;44:553-563. PMID: 5877505.
- Wisniewski H, Terry RD, Hirano A. Neurofibrillary pathology. J Neuropathol Exp Neurol 1970; 29: 163-176. PMID: 5435819.
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120: 885-890. https://doi.org/10.1016/s0006-291x(84)80190-4
- Glenner GG, Wong CW, Quaranta V, et al. The amyloid deposits in Alzheimer's disease: their nature and pathogenesis. Appl Pathol 1984; 12:357-369. PMID: 6242724.
- Masters CL, Simms G, Weinman NA, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Nat Acad Sci USA 1985; 82:4245-4249. https://doi.org/10.1073/pnas.82.12.4245
- Iwatsubo T, Odaka A, Suzuki N, et al. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 1994; 13:45-53. https://doi.org/10.1016/0896-6273(94)90458-8
- Masters CL, Beyreuther K. Amyloid-β production. In: Dickson DW, Weller RO, eds. Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd ed. Blackwell Publishing Co; 2011:92–6.
- Lee MS, Kao SC, Lemere CA, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol 2013;163:83-95. https://doi.org/10.1083/jcb.200301115
- Vassar R, Kovacs DM, Yan R, et al. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential J Neurosci 2009;29:12787-12794. https://doi.org/10.1523/JNEUROSCI.3657-09.2009
- Zhang X, Li Y, Xu H, Zhang YW. The gamma-secretase complex: from structure to function. Front Cell Neurosci 2014;8:427. https://doi.org/10.3389/fncel.2014.00427
- Barthet G, Georgakopoulos A, Robakis NK. Cellular mechanisms of γ-secretase substrate selection, processing and toxicity. Prog Neurobiol 2012;98:166-175. https://doi.org/10.1016/j.pneurobio.2012.05.006
- Wolfe MS. Structure and function of the γ-secretase complex. Biochemistry 2019;58:2953-2966. https://doi.org/10.1021/acs.biochem 9b00401.
- Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer's disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol 1991;1:241-251. https://doi.org/10.1111/j.1750-3639.1991.tb00667.x
- Masters CL, Beyreuther K. Alzheimer's centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 2006;129:2823-2839. https://doi.org/10.1093/brain/awl251
- Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006262. https://doi.org/10.1101/cshperspect.a006262
- Chen GF, Xu TH, Yan Y, et al. Amyloid beta: structure, biology and structure-based therapeutic treatment. Acta Pharmacol Sin 2017;38:1205-1235. https://doi.org/10.1038/aps.2017.28
- Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA 2001;98:5856-5861. https://doi.org/10.1073/pnas.081620098
- Wang H, Kulas JA, Wang C, et al. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc Natl Acad Sci USA 2021;118:e2102191118. https://doi.org/10.1073/pnas.2102191118
- Ricciarelli R, Fedele E. cAMP, cGMP and amyloid β: three ideal partners for memory formation. Trends Neurosci 2018;41:255-266. https://doi.org/10.1016/j.tins.2018.02.001
- Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans 2005;33:1101-1105. https://doi.org/10.1042/BST20051101
- Miners JS, Baig S, Palmer J, et al. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol 2008;18:240-252. https://doi.org/10.1111/j.1750-3639.2008.00132.x
- Love S, Miners S, Palmer J, et al. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front Biosci 2009;14:4778-4792. https://doi.org/10.2741/3567
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 2009;118:103-113. https://doi.org/10.1007/s00401-009-0522-3
- Miners JS, Baig S, Tayler H, et al. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J Neuropathol Exp Neurol 2009;68:902-914. https://doi.org/10.1097/NEN.0b013e3181afe475
- Zlokovic BV, Deane R, Sagare AP, et al. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta-peptide elimination from the brain. J Neurochem 2010;115:1077-1089. https://doi.org/10.1111/j.1471-4159.2010.07002.x
- Pacheco-Quinto J, Clausen D, Pérez-González R, et al. Intracellular metalloprotease activity controls intraneuronal Aβ aggregation and limits secretion of Aβ via exosomes. FASEB J 2019;33:3758-3771. https://doi.org/10.1096/fj.201801319R
- Weller RO, Djuanda E, Yow HY, et al. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 2009;117:1-14. https://doi.org/10.1007/s00401-008-0457-0
- Gouveia-Freitas K, Bastos-Leite AJ. Perivascular spaces and brain waste clearance systems: relevance for neurodegenerative and cerebrovascular pathology. Neuroradiology 2021;63:1581-1597. https://doi.org/10.1007/s00234-021-02718-7
- Foster EM, Dangla-Valls A, Lovestone S, et al. Clusterin in Alzheimer's disease: mechanisms, genetics, and lessons from other pathologies. Front Neurosci 2019;13:164. https://doi.org/10.3389/fnins.2019.00164
- Baig S, Palmer LE, Owen MJ, et al. Clusterin mRNA and protein in Alzheimer's disease. J Alzheimers Dis 2012;28:337-344. https://doi.org/10.3233/JAD-2011-110473
- Miners JS, Clarke P, Love S. Clusterin levels are increased in Alzheimer's disease and influence the regional distribution of Aβ. Brain Pathol 2017;27:305-313. https://doi.org/10.1111/bpa.12392
- Kida E, Choi-Miura NH, Wisniewski KE. Deposition of apolipoproteins E and J in senile plaques is topographically determined in both Alzheimer's disease and Down's syndrome brain. Brain Res 1995;685:211-216. https://doi.org/10.1016/0006-8993(95)00482-6
- Howlett DR, Hortobágyi T, Francis PT. Clusterin associates specifically with Aβ1-40 in Alzheimer’s disease brain tissue. Brain Pathol 2013;23:623-632. https://doi.org/10.1111/bpa.12057
- Humphreys DT, Carver JA, Easterbrook-Smith SB, et al. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem 1999; 274: 6875-6881. https://doi.org/10.1074/jbc.274.11.6875
- Oh SB, Kim MS, Park S, et al. Clusterin contributes to early stage of Alzheimer's disease pathogenesis. Brain Pathol 2019;29:217-231. https://doi.org/10.1111/bpa.12660
- Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 2007;27: 909-918. https://doi.org/10.1038/sj.jcbfm.9600419
- Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol 2015;11:457-470. https://doi.org/10.1038/nrneurol.2015.119
- Calero M, Rostagno A, Matsubara E, et al. Apolipoprotein J (clusterin) and Alzheimer's disease. Microsc Res Tech 2000;50:305-315. https://doi.org/10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L
- Matsubara E, Soto C, Governale S, Frangione B, Ghiso J. Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem J 1996;316:671-679. https://doi.org/10.1042/bj3160671
- Robbins JP, Perfect L, Ribe EM, et al. Clusterin is required for β-amyloid toxicity in human iPSC-derived neurons. Front Neurosci 2018;12:504. https://doi.org/10.3389/fnins.2018.00504
- Zhou Y, Hayashi I, Wong J, et al. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer's disease. PLoS One 2014;9:e103187. https://doi.org/10.1371/journal.pone.0103187
- Rijal Upadhaya A, Kosterin I, Kumar S, et al. Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer's disease. Brain 2014;137:887-903. https://doi.org/10.1093/brain/awt362
- Thal DR, Walter J, Saido TC, Fändrich M. Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer's disease. Acta Neuropathol 2015;129:167-182. https://doi.org/10.1007/s00401-014-1375-y
- Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998;95:6448-53. https://doi.org/10.1073/pnas.95.11.6448
- Gong Y, Chang L, Viola KL, et al. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 2003;100:10417-10422. https://doi.org/10.1073/pnas.1834302100
- Hayden EY, Teplow DB. Amyloid beta-protein oligomers and Alzheimer’s disease, Alzheimers Res Ther 2013; 5: 60. https://doi.org/10.1186/alzrt226
- Viola KL, Klein WL. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 2015;129:183-206. https://doi.org/10.1007/s00401-015-1386-3
- Mroczko B, Groblewska M, Litman-Zawadzka A, et al. Cellular receptors of amyloid β oligomers (AβOs) in Alzheimer's disease. Int J Mol Sci 2018;19:1884. https://doi.org/10.3390/ijms19071884
- Mroczko B, Groblewska M, Litman-Zawadzka A, et al. Amyloid β oligomers (AβOs) in Alzheimer's disease. J Neural Transm 2018;125:177-191. https://doi.org/10.1007/s00702-017-1820-x
- Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003;300:486-489. https://doi.org/10.1126/science.1079469
- Deshpande A, Mina E, Glabe C, et al. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 2006;26:6011-6018. https://doi.org/10.1523/JNEUROSCI.1189-06.2006
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 2006;27:570-575. https://doi.org/10.1016/j.neurobiolaging.2005.04.017
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem 2007;101:1172-1184. https://doi.org/10.1111/j.1471-4159.2006.04426.x
- Glabe CG, Kayed R. common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006;66:S74-8. https://doi.org/10.1212/01.wnl.0000192103.24796.42
- Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837-842. https://doi.org/10.1038/nm1782
- Sengupta U, Nilson AN, Kayed R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016;6:42-49. https://doi.org/10.1016/j.ebiom.2016.03.035
- Fukumoto H, Tokuda T, Kasai T, et al. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J 2010;24:2716-2726. https://doi.org/10.1096/fj.09-150359
- Esparza TJ, Zhao H, Cirrito JR, et al. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol 2013;73:104-119. https://doi.org/10.1002/ana.23748
- Del Río JA, Ferrer I, Gavín R. Role of celular prion protein in interneuronal amyloid transmission. Prog Neurobiol 2018;165-167:87-102. https://doi.org/10.1016/j.pneurobio.2018.03.001
- Gunther EC, Strittmatter SM. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J Mol. Med 2010; 88: 331-338. https://doi.org/10.1007/s00109-009-0568-7
- Dohler F, Sepulveda-Falla D, Krasemann S, et al. High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014; 137: 873-886. https://doi.org/10.1093/brain/awt375
- Fluharty BR, Biasini E, Stravalaci M, et al. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem 2013;288:7857-7866. https://doi.org/10.1074/jbc.M112.423954
- Freir DB, Nicoll AJ, Klyubin I, et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011;2:336. https://doi.org/10.1038/ncomms1341
- Ganzinger KA, Narayan P, Qamar SS, et al. Single-molecule imaging reveals that small amyloid-beta1-42 oligomers interact with the cellular prion protein (PrP(C)). Chembiochem 2014;15:2515-2521. https://doi.org/10.1002/cbic.201402377
- Lauren J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128-1132. https://doi.org/10.1038/nature07761
- Hartmann A, Muth C, Dabrowski O, et al. Exosomes and the prion protein: more than one truth. Front Neurosci 2017;11:194. https://doi.org/10.3389/fnins.2017.00194
- Chaudhary H, Meister SW, Zetterberg H, et al. Dissecting the structural organization of multiprotein amyloid aggregates using a bottom-up approach. ACS Chem Neurosci 2020;11:1447-1457. https://doi.org/10.1021/acschemneuro.0c00110
- Rahman MM, Lendel C. Extracellular protein components of amyloid plaques and their roles in Alzheimer's disease pathology. Mol Neurodegener 2021;16:59. https://doi.org/10.1186/s13024-021-00465-0
- Ferrer I, Blanco R, Carmona M, et al. Prion protein expression in senile plaques in Alzheimer's disease. Acta Neuropathol 2001;101:49-56. https://doi.org/10.1007/s004010000271
- Ferrer I, Martí E, Tortosa A, et al. Dystrophic neurites of senile plaques are defective in proteins involved in exocytosis and neurotransmission. J Neuropathol Exp Neurol 1998;57:218-225. https://doi.org/10.1097/00005072-199803000-00002
- Lenders MB, Peers MC, Tramu G, et al. Dystrophic peptidergic neurites in senile plaques of Alzheimer's disease hippocampus precede formation of paired helical filaments. Brain Res 1989;481:344-349. https://doi.org/10.1016/0006-8993(89)90812-3
- Benzing WC, Brady DR, Mufson EJ, et al. Evidence that transmitter-containing dystrophic neurites precede those containing paired helical filaments within senile plaques in the entorhinal cortex of nondemented elderly and Alzheimer's disease patients. Brain Res 1993;619:55-68. https://doi.org/10.1016/0006-8993(93)91595-j
- Benzing WC, Ikonomovic MD, Brady DR, et al. Evidence that transmitter-containing dystrophic neurites precede paired helical filament and Alz-50 formation within senile plaques in the amygdala of nondemented elderly and patients with Alzheimer's disease. J Comp Neurol 1993;334:176-191. https://doi.org/10.1002/cne.903340203
- Dickson DW, Farlo J, Davies P, et al. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 1988;132:86-101. PMID: 2456021.
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15:267-278. https://doi.org/10.1515/revneuro 2004.15.4.267.
- Brion JP, Couck AM, Bruce M, et al. Synaptophysin and chromogranin A immunoreactivities in senile plaques of Alzheimer's disease. Brain Res 1991;539:143-150. https://doi.org/10.1016/0006-8993(91)90697-t
- Munoz DG. Chromogranin A-like immunoreactive neurites are major constituents of senile plaques. Lab Invest 1991;64:826-832. PMID: 1710735.
- Marksteiner J, Kaufmann WA, Gurka P, et al. Synaptic proteins in Alzheimer's disease. J Mol Neurosci 2002;18:53-63. https://doi.org/10.1385/JMN:18:1-2:53
- Barranco N, Plá V, Alcolea D, et https://doi.org/10.1385/JMN:18:1-2:53.al Dense core vesicle markers in CSF and cortical tissues of patients with Alzheimer's disease. Transl Neurodegener 2021;10:37. https://doi.org/10.1186/s40035-021-00263-0
- Andrés-Benito P, Carmona M, Jordán Pirla M, et al. Dysregulated protein phosphorylation as main contributor of granulovacuolar degeneration at the first stages of NFT pathology. Neuroscience 2021 Oct 30;S0306-4522(21)00534-0. https://doi.org/10.1016/j.neuroscience.2021.10.023
- Woodhouse A, Vickers JC, Dickson TC. Cytoplasmic cytochrome c immunolabelling in dystrophic neurites in Alzheimer's disease. Acta Neuropathol 2006;112:429-437. https://doi.org/10.1007/s00401-006-0107-3
- Pérez-Gracia E, Torrejón-Escribano B, Ferrer I. Dystrophic neurites of senile plaques in Alzheimer's disease are deficient in cytochrome c oxidase. https://doi.org/10.1007/s00401-008-0370-6
- Armstrong DM, Bruce G, Hersh LB, et al. Choline acetyltransferase immunoreactivity in neuritic plaques of Alzheimer brain. Neurosci Lett 1986;71:229-234. https://doi.org/10.1016/0304-3940(86)90564-1
- Masliah E, Mallory M, DeTeresa R, et al. Differing patterns of aberrant neuronal sprouting in Alzheimer's disease with and without Lewy bodies. Brain Res 1993;617:258-266. https://doi.org/10.1016/0006-8993(93)91093-8
- Ferrer I, Zújar MJ, Rivera R, et al. Parvalmunin-immunoreactive dystrophic neurites and aberrant sprouts in the cerebral cortex of patients with Alzheimer’s disease. Neurosci Lett 1993;158:163-166. https://doi.org/10.1016/0304-3940(93)90254-i
- Adams LA, Munoz DG. Differential incorporation of processes derived from different classes of neurons into senile plaques in Alzheimer's disease. Acta Neuropathol 1993;86:365-370. https://doi.org/10.1007/BF00369449
- Dickson TC, King CE, McCormack GH, et al. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp Neurol 1999;156:100-110. https://doi.org/10.1006/exnr.1998.7010
- Adlard PA, Vickers JC. Morphologically distinct plaque-types differentially affect dendritic structure and organisation in the early and late stages of Alzheimer’s disease. Acta Neuropathol 2002;103:377-383. https://doi.org/10.1007/s00401-001-0476-6
- Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 2005;25:7278-7287. https://doi.org/10.1523/JNEUROSCI.1879-05.2005
- Knafo S, Alonso-Nanclares L, Gonzalez-Soriano J, et al. Widespread changes in dendritic spines in a model of Alzheimer's disease. Cereb Cortex. 2009;19:586-592. https://doi.org/10.1093/cercor/bhn111
- Robbins M, Clayton E, Kaminski Schierle GS. Synaptic tau: A pathological or physiological phenomenon? Acta Neuropathol Commun 2021;9:149. https://doi.org/10.1186/s40478-021-01246-y
- Alquezar C, Arya S, Kao AW. Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 2021;11:595532. https://doi.org/10.3389/fneur.2020.595532
- Hernández F, Avila J. Intra- and extracellular protein interactions with tau. Curr Alzheimer Res 2010;7:670-676. https://doi.org/10.2174/156720510793611583
- Rauch JN, Olson SH, Gestwicki JE. Interactions between microtubule-associated protein tau (MAPT) and small molecules. Cold Spring Harb Perspect Med 2017;7:a024034. https://doi.org/10.1101/cshperspect.a024034
- Borna H, Assadoulahei K, Riazi G, et al. Structure, function and interactions of tau: particular focus on potential drug targets for the treatment of tauopathies. CNS Neurol Disord Drug Targets 2018;17:325-337. https://doi.org/10.2174/1871527317666180525112008
- Trushina NI, Bakota L, Mulkidjanian AY, et al. The evolution of tau phosphorylation and interactions. Front Aging Neurosci 2019;11:256. https://doi.org/10.3389/fnagi.2019.00256
- Brandt R, Trushina NI, Bakota L. Much more than a cytoskeletal protein: physiological and pathological functions of the non-microtubule binding region of tau. Front Neurol 2020;11:590059. https://doi.org/10.3389/fneur.2020.590059
- Sinsky J, Pichlerova K, Hanes J. Tau protein interaction partners and their roles in Alzheimer's disease and other tauopathies. Int J Mol Sci 2021;22:9207. https://doi.org/10.3390/ijms22179207
- Delacourte A, Defossez A. Alzheimer's disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurol Sci 1986;76:173-186. https://doi.org/10.1016/0022-510x(86)90167-x
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA 1986;83:4044-4048. https://doi.org/10.1073/pnas.83.11.4044
- Wood JG, Mirra SS, Pollock NJ, et al. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc Natl Acad Sci USA 1986;83:4040-4043. https://doi.org/10.1073/pnas.83.11.4040
- Grundke-Iqbal I, Iqbal K, Quinlan M, et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 1986;261: 6084-6089. PMID: 3084478.
- Iqbal K, Grundke-Iqbal I, Wisniewski HM. Neuronal cytoskeleton in aging and dementia. Prog Brain Res 1986;70:279-288. https://doi.org/10.1016/s0079-6123(08)64310-1
- Goedert M, Wischik CM, Crowther RA, et al. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as microtubule-associated protein tau. Proc Natl Acad Sci USA 1988;85: 4051-4055. https://doi.org/10.1073/pnas.85.11.4051
- Wischik CM, Novak M, Edwards PC, et al. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA 1988;85:4884-4888. https://doi.org/10.1073/pnas.85.13.4884
- Kosik KS, Orecchio LD, Binder L, et al. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1988;1:817-825. https://doi.org/10.1016/0896-6273(88)90129-8
- Delacourte A, Buée L. Normal and pathological tau proteins as factors of microtubule assembly. Int Rev Cytol 1997;171:167-224. https://doi.org/10.1016/s0074-7696(08)62588-7
- Buée L, Bussière T, Buée-Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000; 33:95-130. https://doi.org/10.1016/s0165-0173(00)00019-9
- Iqbal K, Grundke-Iqbal I. Discoveries of tau, abnormally hyperphosphorylated tau and others of neurofibrillary degeneration: A personal historical perspective. J Alzheimers Dis 2006;9:219-242. https://doi.org/10.3233/jad-2006-9s325
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofbrillary degeneration. Cold Spring Harb Perspect Med 2012;2: a006247. https://doi.org/10.1101/cshperspect.a006247
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol 2013;12: 609-622. https://doi.org/10.1016/S1474-4422(13)70090-5
- Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull 2016, 26: 238-292. https://doi.org/10.1016/j.brainresbull.2016.08.018
- Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 2016;12:15-27. https://doi.org/10.1038/nrneurol.2015.225
- Goedert M, Spillantini MG. Ordered Assembly of Tau Protein and Neurodegeneration. Adv Exp Med Biol 2019;1184:3-21. https://doi.org/10.1007/978-981-32-9358-8_1
- Goedert M, Spillantini MG, Cairns NJ, et al. Tau proteins in Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 1992; 8: 159-168. https://doi.org/10.1016/0896-6273(92)90117-v
- Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999;52: 1158-1165. https://doi.org/10.1212/wnl.52.6.1158
- Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. Embo J 1993;12:365-370. https://doi.org/10.1002/j.1460-2075.1993.tb05665.x
- Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res 1999; 55:713-723. https://doi.org/10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G
- Weaver CL, Espinoza M, Kress Y, et al. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 2000;21:719-727. https://doi.org/10.1016/s0197-4580(00)00157-3
- Gamblin TC, Chen F, Zambrano A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA 2003;100:10032-10037. https://doi.org/10.1073/pnas.1630428100
- García-Sierra F, Ghoshal N, Quinn B, et al. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis 2003;5:65-77. https://doi.org/10.3233/jad-2003-5201
- Rissman RA, Poon WW, Blurton-Jones M, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 2004;114:121-130. https://doi.org/10.1172/JCI20640
- Hyman BT, Augustinack JC, Ingelsson M. Transcriptional and conformational changes of the tau molecule in Alzheimer's disease. Biochim Biophys Acta 2005;1739:150-157. https://doi.org/10.1016/j.bbadis.2004.06.015
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 2005;1739:91-103. https://doi.org/10.1016/j.bbadis.2004.08.010
- Avila J. Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett 2006;580:2922-2927. https://doi.org/10.1016/j.febslet.2006.02.067
- Zilka N, Filipcik P, Koson P, et al. Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 2006;580:3582-3588. https://doi.org/10.1016/j.febslet.2006.05.029
- Luna-Muñoz J, Chávez-Macías L, García-Sierra F, et al. Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer's disease. J Alzheimers Dis 2007;12:365-375. https://doi.org/10.3233/jad-2007-12410
- García-Sierra F, Mondragón-Rodríguez S, Basurto-Islas G. Truncation of tau protein and its pathological significance in Alzheimer's disease. https://doi.org/10.3233/jad-2008-14407
- Mondragón-Rodríguez S, Basurto-Islas G, Santa-Maria I, et al. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer's disease. Int J Exp Pathol 2008;89:81-90. https://doi.org/10.1111/j.1365-2613.2007.00568.x
- Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int 2011;58:458-471. https://doi.org/10.1016/j.neuint.2010.12.023
- Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2011;2:252. https://doi.org/10.1038/ncomms1255
- Takashima A. Tauopathies and tau oligomers. J Alzheimers Dis 2013;37:565-568. https://doi.org/10.3233/JAD-130653
- Hernández F, Pérez M, Gómez de Barreda E et al. Tau as a molecular marker of development, aging and neurodegenerative disorders. Curr Aging Sci 2008;1:56-61. https://doi.org/10.2174/1874609810801010056
- Fitzpatrick AWP, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 2017; 547:185-190. https://doi.org/10.1038/nature23002
- Ramesh M, Gopinath P, Govindaraju T. Role of Post-translational Modifications in Alzheimer's disease. Chembiochem 2020;21:1052-1079. https://doi.org/10.1002/cbic.201900573
- Wang D, Huang X, Yan L, et al. The structure biology of tau and clue for aggregation inhibitor design. Protein J 2021;40:656-668. https://doi.org/10.1007/s10930-021-10017-6
- Moloney CM, Lowe VJ, Murray ME. Visualization of neurofibrillary tangle maturity in Alzheimer's disease: A clinicopathologic perspective for biomarker research. Alzheimers Dement 2021;17:1554-1574. https://doi.org/10.1002/alz.12321
- Ferrer I, López-González I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 2014;73:81-97. https://doi.org/10.1097/NEN.0000000000000030
- Kametani F, Yoshida M, Matsubara T, et al. Comparison of common and disease-specific post-translational modifications of pathological tau associated with a wide range of tauopathies. Front Neurosci 2020; 14: 581936. https://doi.org/10.3389/fnins.2020.581936
- Wang JZ, Xia YY, Grundke-Iqbal I, et al. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 2013;33:S123-139. https://doi.org/10.3233/JAD-2012-129031
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med 2009;15:112-119. https://doi.org/10.1016/j.molmed.2009.01.003
- Ferrer I, Blanco R, Carmona M, et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 2001;11:144-158. https://doi.org/10.1111/j.1750-3639.2001.tb00387.x
- Ferrer I, Blanco R, Carmona M, et al. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J Neural Transm 2001;108:1397-415. https://doi.org/10.1007/s007020100016
- Ferrer I, Gomez-Isla T, Puig B, et al. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res 2005;2:3-18. https://doi.org/10.2174/1567205052772713
- Guimaraes TR, Swanson E, Kofler J, et al. G-protein-coupled receptor kinases are associated with Alzheimer’s disease pathology. Neuropathol Appl Neurobiol 2021;47:942-957. https://doi.org/10.1111/nan.12742
- Cowan CM, Quraishe S, Mudher A What is the pathological significance of tau oligomers? Biochem Soc Trans 2012;40:693-697. https://doi.org/10.1042/BST20120135
- Moraga DM, Nunez P, Garrido J, et al. A tau fragment containing a repetitive sequence induces bundling of actin filaments. J Neurochem 1993;61:979-986. https://doi.org/10.1111/j.1471-4159.1993 tb03611.x.
- Maas T, Eidenmüller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 2000;275:15733-15740. https://doi.org/10.1074/jbc.M00038920
- Shea TB, Chan A. S-adenosyl methionine: a natural therapeutic agent effective against multiple hallmarks and risk factors associated with Alzheimer's disease. J Alzheimers Dis 2008;13:67-70. https://doi.org/10.3233/jad-2008-13107
- Schneider A, Biernat J, von Bergen M, et al. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999;38:3549-3558. https://doi.org/10.1021/bi981874p
- Farias GA, Muñoz JP, Garrido J, et al. Tubulin, actin, and tau protein interactions and the study of their macromolecular assemblies. J Cell Biochem 2002;85:315-324. https://doi.org/10.1002/jcb.10133
- Mohan R, John A. Microtubule-associated proteins as direct crosslinkers of actin filaments and microtubules. IUBMB Life 2015;67:395-403. https://doi.org/10.1002/iub.1384
- Sallaberry CA, Voss BJ, Majewski J, et al. Tau and membranes: interactions that promote folding and condensation. Front Cell Develop Biol 2021;9:725241. https://doi.org/10.3389/fcell.2021.725241
- Brunello CA, Merezhko M, Uronen RL, et al. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci 2020;77:1721-1744. https://doi.org/10.1007/s00018-019-03349-1
- Bok E, Leem E, Lee BR, et al. Role of the lipid membrane and membrane proteins in tau pathology. Front Cell Dev Biol 2021;9:653815. https://doi.org/10.3389/fcell.2021.653815
- Annadurai N, De Sanctis JB, Hajdúch M, et al. Tau secretion and propagation: Perspectives for potential preventive interventions in Alzheimer's disease and other tauopathies. Exp Neurol 2021;343:113756. https://doi.org/10.1016/j.expneurol.2021.113756
- Santa-Maria I, Varghese M, Ksiéżak-Reding H, et al. Paired helical filaments from Alzheimer disease brain induces intracellular accumulation of Tau protein in aggresomes. J Biol Chem 2012: 287: 20522- 20533. https://doi.org/10.1074/jbc.M111.323279
- Falcon B, Zhang W, Schweighauser M, et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol 2018;136:699-708. https://doi.org/10.1007/s00401-018-1914-z
- Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature 2018;561:137-140. https://doi.org/10.1038/s41586-018-0454-y
- Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019;568:420-423. https://doi.org/10.1038/s41586-019-1026-5
- Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature 2020;580:283-287. https://doi.org/10.1038/s41586-020-2043-0
- Arakhamia T, Lee CE, Carlomagno Y, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 2020;180:633-644.e12. https://doi.org/10.1016/j.cell.2020.01.027
- Shi Y, Murzin AG, Falcon B, et al. Cryo-EM structures of tau filaments from Alzheimer's disease with PET ligand APN-1607. Acta Neuropathol 2021;141: 697-708. https://doi.org/10.1007/s00401-021-02294-3
- Shi Y, Zhang W, Yang Y, et al. Structure-based classification of tauopathies. Nature 2021;598:359-363. https://doi.org/10.1038/s41586-021-03911-7
- Smith R, Strandberg O, Mattsson-Carlgren N, et al. The accumulation rate of tau aggregates is higher in females and younger amyloid-positive subjects. Brain 2020;143:3805-3815. https://doi.org/10.1093/brain/awaa327
- Davis EJ, CarolineW. Solsberg CW, et al. Sex-specific association of the X chromosome with cognitive change and tau pathology in aging and Alzheimer disease. JAMA Neurol 2021; 78:1249-1254. https://doi.org/10.1001/jamaneurol.2021.2806
- Meier S, Bell M, Lyons DN, et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer’s disease brain. J Alzheimers Dis 2015; 48: 687-702. https://doi.org/10.3233/JAD-150298
- Ayyadevara S, Balasubramaniam M, Parcon PA, et al. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer’s hippocampus from normal controls. Aging Cell 2016; 15: 924-939. https://doi.org/10.1111/acel.12501
- Drummond E, Pires G, MacMurray C, et al. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020;143:2803-2817. https://doi.org/10.1093/brain/awaa223
- van Leeuwen FW, Fischer DF, Kamel D, et al. Molecular misreading: a new type of transcript mutation expressed during aging. Neurobiol Aging 2000;21:879-891. https://doi.org/10.1016/s0197-4580(00)00151-2
- van Leeuwen FW, Hol EM, Fischer DF. Frameshift proteins in Alzheimer's disease and in other conformational disorders: time for the ubiquitin-proteasome system. J Alzheimers Dis 2006;9:319-325. https://doi.org/10.3233/jad-2006-9s336
- Vergara C, Ordonez-Gutierrez L, Wandosell F, et al. Role of PrP(C) expression in tau protein levels and phosphorylation in Alzheimer’s disease evolution. Mol Neurobiol 2015;51:1206-1220. https://doi.org/10.1007/s12035-014-8793-7
- Chen RJ, Chang WW, Lin YC, et al. Alzheimer’s amyloid-beta oligomers rescue cellular prion protein induced tau reduction via Fyn pathways. ACS Chem Neurosci 2013; 4:1287-1296. https://doi.org/10.1021/cn400085q
- Schmitz M, Wulf K, Signore SC, et al. Impact of the cellular prion protein on amyloid-beta and 3PO-tau processing. J Alzheimers Dis 2014;38: 551-565. https://doi.org/10.3233/JAD-130566
- Lidón L, Vergara C, Ferrer I, et al. Tau protein as a new regulator of cellular prion protein transcription. Mol Neurobiol 2020;57:4170-4186. https://doi.org/10.1007/s12035-020-02025-x
- Lidón L, Llaó-Hierro L, Nuvolone M, et al. Tau exon 10 inclusion by PrPC through down-regulating GSK3β activity. Int J Mol Sci 2021;22: 5370. https://doi.org/10.3390/ijms22105370
- Gavín R, Lidón L, Ferrer I, Del Río JA. The quest for cellular prion protein functions in the aged and neurodegenerating brain. Cells 2020;9:591. https://doi.org/10.3390/cells9030591
- Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991;349:704-706. https://doi.org/10.1038/349704a0
- Chartier-Harlin MC, Crawford F, Houlden H, et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 1991; 353: 844-846. https://doi.org/10.1038/353844a0
- Murrell J, Farlow M, Ghetti B, et al. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 1991;254:97-99. https://doi.org/10.1126/science.1925564
- Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995; 18;269:973-977. https://doi.org/10.1126/science.7638622
- Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995;375:754-760. https://doi.org/10.1038/375754a0
- Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 1995;376:775-778. https://doi.org/10.1038/376775a0
- Goedert M, Spillantini MG. A Century of Alzheimer’s disease. Science 2006; 314: 777-781. https://doi.org/10.1126/science.1132814
- Bertram L, Tanzi RE. Genetics of Alzheimer’s disease. In: Dickson DW, Weller RO (eds) The molecular pathology of dementias and movement disorders, 2nd edn. Wiley-Blackwell, Oxford, 2011; pp 51-61
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256:184-185. https://doi.org/10.1126/science.1566067
- Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293:1487-1491. https://doi.org/10.1126/science.1058189
- Ribé EM, Pérez M, Puig B, et al. Accelerated amyloid deposition, neurofibrillary degeneration, and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis 2005;20:814-822. https://doi.org/10.1016/j.nbd.2005.05.027
- Samura E, Shoji M, Kawarabayashi T, et al. Enhanced accumulation of tau in doubly transgenic mice expressing mutant betaAPP and presenilin-1. Brain Res 2006;1094:192-199. https://doi.org/10.1016/j.brainres.2005.12.134
- Bennett RE, DeVos SL, Dujardin S, et al. Enhanced tau aggregation in the presence of amyloid β. Am J Pathol 2017;187:1601-1612. https://doi.org/10.1016/j.ajpath.2017.03.011
- Hyman BT, West HL, Rebeck GW, et al. Neuropathological changes in Down's syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol 1995; 52:373-378. https://doi.org/10.1001/archneur.1995.00540280059019
- Hof PR, Bouras C, Perl DP, et al. Age-related distribution of neuropathological changes in the cerebral cortex of patients with Down's syndrome. Arch Neurol 1995; 52:379-391. https://doi.org/10.1001/archneur.1995.00540280065020
- Lemere CA, Blusztajn JK, Yamaguchi H, et al. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 1996;3:16-32. https://doi.org/10.1006/nbdi.1996.0003
- Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement 2015;11:700-709. https://doi.org/10.1016/j.jalz.2014.10.007
- Tu S, Okamoto S, Lipton SA, et al. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener 2014;9:48. https://doi.org/10.1186/1750-1326-9-48
- Hillen H. The beta-amyloid dysfunction (BAD) hypothesis for Alzheimer’s disease. Front Neurosci 2019; 13:1154. https://doi.org/10.3389/fnins.2019.01154
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016; 8:595-608. https://doi.org/10.15252/emmm.201606210
- Cline EN, Bicca MA, Viola KL, et al. The Amyloid-β oligomer hypothesis: beginning of the third decade. J Alzheimers Dis 2018;64:S567-S610. https://doi.org/10.3233/JAD-179941
- van Helmond Z, Scott Miners J, Kehoe PG, et al. Higher soluble amyloid beta concentration in frontal cortex of young adults than in normal elderly or Alzheimer's disease. Brain Pathol 2010;20:787-793. https://doi.org/10.1111/j.1750-3639.2010.00374.x
- Miners JS, Jones R, Love S. Differential changes in Aβ42 and Aβ40 with age. J Alzheimers Dis 2014;40:727-735. https://doi.org/10.3233/JAD-132339
- van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 2020;21:21-35. https://doi.org/10.1038/s41583-019-0240-3
- Ochalek A, Mihalik B, Avci HX, et al. Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther 2017;9:90. https://doi.org/10.1186/s13195-017-0317-z
- Holton JL, Ghiso J, Lashley T, et al. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol 2001;158:515-526. https://doi.org/10.1016/S0002-9440(10)63993-4
- Garringer HJ, Murrell J, Sammeta N, et al. Increased tau phosphorylation and tau truncation, and decreased synaptophysin levels in mutant BRI2/tau transgenic mice. PLoS One 2013;8:e56426. https://doi.org/10.1371/journal.pone.0056426
- Alzualde A, Indakoetxea B, Ferrer I, et al. A novel PRNP Y218N mutation in Gerstmann–Sträussler–Scheinker disease with neurofibrillary degeneration J Neuropathol Exp Neurol 2010; 69: 789-800. https://doi.org/10.1097/NEN.0b013e3181e85737
- Ghetti B, Piccardo P, Zanusso G. Dominantly inherited prion protein cerebral amyloidoses - a modern view of Gerstmann-Sträussler-Scheinker. Handb Clin Neurol 2018;153:243-269. https://doi.org/10.1016/B978-0-444-63945-5.00014-3
- Hallinan GI, Hoq MR, Ghosh M, et al. Structure of Tau filaments in Prion protein amyloidoses. Acta Neuropathol 2021;142:227-241. https://doi.org/10.1007/s00401-021-02336-w
- Gibson G, Gunasekera N, Lee M, et al. Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. J Neurochem 2004;88:281-290. https://doi.org/10.1046/j.1471-4159.2003.02134.x
- Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261, 921–923. https://doi.org/10.1126/science.8346443
- Saunders AM, Strittmatter WJ, Schmechel D, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 1993; 43: 1467-1472. https://doi.org/10.1212/wnl.43.8.1467
- Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 1977-1981. https://doi.org/10.1073/pnas.90.5.1977
- Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 27, 1349-1356. PMID: 9343467
- Harold D, Abraham R, Hollingworth P, et al. Genome-wide association studies identifies variants CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009;41:1088-1093. https://doi.org/10.1038/ng.440
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094-1099. https://doi.org/10.1038/ng.439
- Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheiomer’s disease. JAMA 2010;303:1832-1840. https://doi.org/10.1001/jama.2010.574
- Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 2010;67:1473-1484. https://doi.org/10.1001/archneurol.2010.201
- Jones L, Holmans PA, Hamshere ML, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One. 2010;5:e13950. https://doi.org/10.1371/journal.pone.0013950
- Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011;43:429-435. https://doi.org/10.1038/ng.803
- Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452-1458. https://doi.org/10.1038/ng.2802
- Naj AC, Jun G, Reitz C, et al. Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol 2014;71:1394-1404. https://doi.org/10.1001/jamaneurol.2014.1491
- Sims R, van der Lee SJ, Naj AC et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet 2017;49:1373-1384. https://doi.org/10.1038/ng.3916
- Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer's disease. Transl Psychiat 2018; 8: 99. https://doi.org/10.1038/s41398-018-0150-6
- Pimenova AA, Raj T, Goate AM. Untangling genetic risk for Alzheimer's disease. Biol Psychiat 2018; 83: 300-310. https://doi.org/10.1016/j.biopsych.2017.05.014
- Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 2019; 51: 404-413. https://doi.org/10.1038/s41588-018-0311-9
- Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019; 51: 414-430. https://doi.org/10.1038/s41588-019-0358-2
- Andrews SJ, Fulton-Howard B, Goate A. Interpretation of risk loci from genome-wide association studies of Alzheimer's disease. Lancet Neurol 2020;19:326-335. https://doi.org/10.1016/S1474-4422(19)30435-1
- Tansey KE, Cameron D, Hill MJ. Genetic risk for Alzheimer's disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med 2018;10:14. https://doi.org/10.1186/s13073-018-0523-8
- Bellenguez C, Grenier-Boley B, Lambert JC. Genetics of Alzheimer's disease: where we are, and where we are going. Curr Opin Neurobiol 2020;61:40-48. https://doi.org/10.1016/j.conb.2019.11.024
- Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 2022;54:412-436. https://doi.org/10.1038/s41588-022-01024-z
- Vogt BA, Vogt LJ, Hof PR. Patterns of cortical neurodegeneration in Alzheimer’s disease : subgroups, subtypes, and implications for staging strategies. In Functional Neurobiology of Aging. Hof PR, Mobbs CV (eds). Academic Press 2001, pp: 111-129
- Hof PR, Cox K, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer's disease: I. Superior frontal and inferior temporal cortex. J Comp Neurol 1990;301:44-54. https://doi.org/10.1002/cne.903010105
- Hof PR, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer's disease: II. Primary and secondary visual cortex. J Comp Neurol 1990;30:55-64. https://doi.org/10.1002/cne.903010106
- Morrison JH, Lewis DA, Campbell MJ, et al. A monoclonal antibody to non-phosphorylated neurofilament protein marks the vulnerable cortical neurons in Alzheimer's disease. Brain Res 1987;416:331-336. https://doi.org/10.1016/0006-8993(87)90914-0
- Vickers JC, Riederer BM, Marugg RA, et al. Alterations in neurofilament protein immunoreactivity in human hippocampal neurons related to normal aging and Alzheimer's disease. Neuroscience 1994;62:1-13. https://doi.org/10.1016/0306-4522(94)90310-7
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science 1997;278:412-419. https://doi.org/10.1126/science.278.5337 412.
- Vickers JC, Dickson TC, Adlard PA, et al. The cause of neuronal degeneration in Alzheimer's disease. Prog Neurobiol 2000;60:139-165. https://doi.org/10.1016/s0301-0082(99)00023-4
- Chan-Palay V. Somatostatin immunoreactive neurons in the human hippocampus and cortex shown by immunogold/silver intensification on vibratome sections: coexistence with neuropeptide Y neurons, and effects in Alzheimer-type dementia. J Comp Neurol 1987; 260: 201-223. https://doi.org/10.1002/cne.902600205
- Rossor MN, Emson PC, Mountjoy CQ, et al. Reduced amounts of immunoreactive somatostatin in the temporal cortex in senile dementia of Alzheimer type. Neurosci Lett 1980; 20: 373-377. https://doi.org/10.1016/0304-3940(80)90177-9
- Rossor MN, Garrett NJ, Johnson AL, et al. A post-mortem study of the cholinergic and GABA systems in senile dementia. Brain 1982;105:313-330. https://doi.org/10.1093/brain/105.2.313
- Hardy J, Cowburn R, Barton A, et al. A disorder of cortical GABAergic innervation in Alzheimer's disease. Neurosci Lett 1987;73:192-196. https://doi.org/10.1016/0304-3940(87)90016-4
- Simpson MD, Cross AJ, Slater P, Deakin JF. Loss of cortical GABA uptake sites in Alzheimer's disease. J Neural Transm 1988;71:219-226. https://doi.org/10.1007/BF01245715
- Gabriel SM, Bierer LM, Harotunian V, et al. Widespread deficits in somatostatin but not neuropeptide Y concentrations in Alzheimer's disease cerebral cortex. Neurosci Lett 1993;155:116-120. https://doi.org/10.1016/0304-3940(93)90686-f
- Beal MF, Mazurek MF, Tran VT, et al. Reduced numbers of somatostatin receptors in the cerebral cortex in Alzheimer's disease. Science 1985;229:289-291. https://doi.org/10.1126/science.2861661
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci 1992;15:303-308. https://doi.org/10.1016/0166-2236(92)90081-i
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 2008;321:53-57. https://doi.org/10.1126/science.1149381
- Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 2016;91:260-292. https://doi.org/10.1016/j.neuron.2016.06.033
- Ferrer I, Soriano E, Tuñón T, et al. Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer's disease. J Neurol Sci 1991;106:135-141. https://doi.org/10.1016/0022-510x(91)90250-b
- Hof PR, Cox K, Young WG, et al. Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer's disease. J Neuropathol Exp Neurol 1991;50:451-462. https://doi.org/10.1097/00005072-199107000-00006
- Sampson VL, Morrison JH, Vickers JC. The cellular basis for the relative resistance of parvalbumin and calretinin immunoreactive neocortical neurons to the pathology of Alzheimer's disease. Exp Neurol 1997;145:295-302. https://doi.org/10.1006/exnr.1997.6433
- Leuba G, Kraftsik R, Saini K. Quantitative distribution of parvalbumin, calretinin, and calbindin D-28k immunoreactive neurons in the visual cortex of normal and Alzheimer cases. Exp Neurol 1998;152:278-291. https://doi.org/10.1006/exnr.1998.6838
- Arai H, Emson PC, Mountjoy CQ, et al. Loss of parvalbumin-immunoreactive neurones from cortex in Alzheimer-type dementia. Brain Res 1987;418:164-169. https://doi.org/10.1016/0006-8993(87)90974-7
- Brady DR, Mufson EJ. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer's diseased brain. Neuroscience 1997;80:1113-1125. https://doi.org/10.1016/s0306-4522(97)00068-7
- Fonseca M, Soriano E, Ferrer I, et al. Chandelier cell axons identified by parvalbumin-immunoreactivity in the normal human temporal cortex and in Alzheimer's disease. Neuroscience 1993; 55: 1107-1116. https://doi.org/10.1016/0306-4522(93)90324-9
- Inaguma Y, Shinohara H, Inagaki T, et al. Immunoreactive parvalbumin concentrations in parahippocampal gyrus decrease in patients with Alzheimer's disease. J Neurol Sci 1992;110:57-61. https://doi.org/10.1016/0022-510x(92)90009-a
- Mikkonen M, Alafuzoff I, Tapiola T, et al. Subfield- and layer-specific changes in parvalbumin, calretinin and calbindin-D28K immunoreactivity in the entorhinal cortex in Alzheimer's disease. Neuroscience 1999;92:515-532. https://doi.org/10.1016/s0306-4522(99)00047-0
- Satoh J, Tabira T, Sano M, et al. Parvalbumin-immunoreactive neurons in the human central nervous system are decreased in Alzheimer's disease. Acta Neuropathol 1991; 81:388-395. https://doi.org/10.1007/BF00293459
- Solodkin A, Veldhuizen SD, Van Hoesen GW. Contingent vulnerability of entorhinal parvalbumin-containing neurons in Alzheimer's disease. J Neurosci 1996;16:3311-3321.
- Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol 1991;111:293-301. https://doi.org/10.1523/JNEUROSCI.16-10-03311.1996
- Ferrer I, Tuñon T, Soriano E, et al. Calbindin D-28k immunoreactivity in the temporal neocortex in patients with Alzheimer's disease. Clin Neuropathol 1993;12:53-58. PMID: 8440080
- Fonseca M, Soriano E. Calretinin-immunoreactive neurons in the normal human temporal cortex and in Alzheimer's disease. Brain Res 1995;691:83-91. https://doi.org/10.1016/0006-8993(95)00622-w
- Hof PR, Nimchinsky EA, Celio MR, et al. Calretinin-immunoreactive neocortical interneurons are unaffected in Alzheimer's disease. Neurosci Lett 1993;152:145-148. https://doi.org/10.1016/0304-3940(93)90504-e
- Brion JP, Resibois A. A subset of calretinin-positive neurons are abnormal in Alzheimer's disease. Acta Neuropathol 1994;88:33-43. https://doi.org/10.1007/BF00294357
- West MJ, Coleman PD, Flood DG, et al. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 1994;344:769-772. https://doi.org/10.1016/s0140-6736(94)92338-8
- Gómez-Isla T, Price JL, McKeel DW Jr, et al. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 1996;16:4491-500. https://doi.org/10.1523/JNEUROSCI.16-14-04491.1996
- Simić G, Kostović I, Winblad B, et al. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol 1997;379:482-494. https://doi.org/10.1002 (sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239-259. https://doi.org/10.1007/BF00308809
- Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271-278. https://doi.org/10.1016/0197-4580(95)00021-6
- Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389-404. https://doi.org/10.1007/s00401-006-0127-z
- Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol 2011;121:171-181. https://doi.org/10.1007/s00401-010-0789-4
- Ferrer I. Defining Alzheimer as a common age-related neurodegenerative process not inevitably leading to dementia. Prog Neurobiol 2012; 97: 38-51. https://doi.org/10.1016/j.pneurobio 2012.03.005.
- Tsartsalis S, Aikaterini Xekardaki A, Hof PR, et al. Early Alzheimer-type lesions in cognitively normal subjects. Neurobiol Aging 2018; 62: 34-44. https://doi.org/10.1016/j.neurobiolaging.2017.10.002
- Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 2012; 71:362-381. https://doi.org/10.1097/NEN.0b013e31825018f7
- Liang WS, Dunckley T, Beach TG, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics 2008; 33: 240-256. https://doi.org/10.1152/physiolgenomics.00242.2007
- Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785-796. https://doi.org/10.1016/S1474-4422(11)70156-9
- Jellinger KA. Pathobiological subtypes of Alzheimer disease. Dement Geriatr Cogn Disord 2020a;49:321-333. https://doi.org/10.1159/000508625
- Grinberg LT, Rüb U, Ferretti RE, et al. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathol Appl Neurobiol 2009;35:406-416. https://doi.org/10.1111/j.1365-2990.2009.00997.x
- Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain 2015; 138:2814-2833. https://doi.org/10.1093/brain/awv236
- Arendt T, Bruckner MK, Morawski M, et al. Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol Commun 2015; 3:10. https://doi.org/10.1186/s40478-015-0187-1
- Andrés-Benito P, Fernández-Dueñas V, Carmona M, et al. Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol Appl Neurobiol 2017;43:373-392. https://doi.org/10.1111/nan.12386
- Eser RA, Ehrenberg AJ, Petersen C, et al. Selective vulnerability of brainstem nuclei in distinct tauopathies: a postmortem study. J Neuropathol Exp Neurol 2018;77:149-161. https://doi.org/10.1093/jnen/nlx113
- Matchett BJ, Grinberg LT, Theofilas P, et al. The mechanistic link between selective vulnerability of the locus ceruleus and neurodegeneration in Alzheimer’s disease. Acta Neuropathol 2021;141:631-650. https://doi.org/10.1007/s00401-020-02248-1
- Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex 1991;1:103-116. https://doi.org/10.1093/cercor/1.1.103
- Ferrer I, Andrés-Benito P. White matter alterations in Alzheimer’s disease without concomitant pathologies. Neuropathol Appl Neurobiol 2020;46:654-672. https://doi.org/10.1111/nan.12618
- Thal DR, Rüb U, Orantes M, et al. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791-1800. https://doi.org/10.1212/wnl.58.12.1791
- Arnsten AFT, Datta D, Del Tredici K, et al. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimers Dement 2021;17:115-124. https://doi.org/10.1002/alz.12192
- Armstrong RA, Myers D, Smith CU. The spatial patterns of plaques and tangles in Alzheimer's disease do not support the 'Cascade Hypothesis'. Dementia 1993;4:16-20. https://doi.org/10.1159/000107291
- Delacourte A, Sergeant N, Champain D, et al. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 2002;59:398-407. https://doi.org/10.1212/wnl.59.3.398
- Delacourte A. Alzheimer’s disease: a true tauopathy fueled by amyloid precursor protein dysfunction. In: Hanin I, Cacabelos R, Fisher A (eds) Recent Progress in Alzheimer’s and Parkinson’s Diseases. Taylor & Francis New York, 2005, pp 301–307
- Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci 2020;23:1183-1193. https://doi.org/10.1038/s41593-020-0687-6
- Mudher A, Lovestone S. Alzheimer's disease-do tauists and baptists finally shake hands? Trends Neurosci 2002;25:22-26. https://doi.org/10.1016/s0166-2236(00)02031-2
- Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol 1995;52:81-88. https://doi.org/10.1001/archneur.1995 00540250089017.
- Bancher C, Braak H, Fischer P, et al. Neuropathological staging of Alzheimers lesions and intellectual status in Alzheimer’s disease and Parkinson’s disease patients. Neurosci Lett 1993;162:179-182. https://doi.org/10.1016/0304-3940(93)90590-h
- Hof PR, Morrison JH. Hippocampal and neocortical involvement in normal brain aging and dementia: morphological and neurochemical profile of the vulnerable circuits. J Am Geriatr Soc 1996;44:857-864. https://doi.org/10.1111/j.1532-5415.1996.tb03748.x
- Bancher C, Jellinger K, Lassmann H, et al. Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci 1996;246:137-146. https://doi.org/10.1007/BF02189115
- Duyckaerts C, Bennecib M, Grignon Y, et al. Modeling the relation between neurofibrillary tangles and mental status. Neurobiol Aging 1997;18:267-273. https://doi.org/10.1016/s0197-4580(97)80306-5
- Haroutunian V, Purohit DP, Perl DP, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol 1999;56:713-718. https://doi.org/10.1001/archneur.56.6.713
- Guillozet AL, Weintraub S, Mash DC, et al. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 2003;60:729-736. https://doi.org/10.1001/archneur.60.5.729
- Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 2003;60:1495-500. https://doi.org/10.1212/01.wnl.0000063311.58879.01
- Imhof A, Kövari E, von Gunten A, et al. Morphological substrates of cognitive decline in nonagenarians and centenarians: a new paradigm? J Neurol Sci 2007;257:72-79. https://doi.org/10.1016/j.jns.2007.01.025
- Hof PR. Regional and laminar patterns of selective neuronal vulnerability in Alzheimer’s disease. In: Functional Neurobiology of Aging., Hof PR, Mobbs CV (eds). Academic Press, New York, 2001:95-109
- Frisoni GB, Altomare D, Thal DR, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nature Rev Neurosci 2021;23:53-66. https://doi.org/10.1038/s41583-021-00533-w
- Price DL, Martin LJ, Sisodia SS, et al. Aged non-human primates: an animal model of age-associated neurodegenerative disease. Brain Pathol 1991;1: 287-296. https://doi.org/10.1111/j.1750-3639.1991.tb00672.x
- Giannakopoulos P, Silhol S, Jallageas V, et al. Quantitative analysis of tau protein-immunoreactive accumulations and beta-amyloid protein deposits in the cerebral cortex of the mouse lemur, Microcebus murinus. Acta Neuropathol 1997;94:131-139. https://doi.org/10.1007/s004010050684
- Head E. Neurobiology of the aging dog. Age 2012;33:485-496. https://doi.org/10.1007/s11357-010-9183-3
- Morelli L, Wei L, Amorim A, et al. Cerebrovascular amyloidosis in squirrel monkeys and rhesus monkeys: apolipoprotein E genotype. FEBS Lett 1996; 379:132-134. https://doi.org/10.1016/0014-5793(95)01491-8
- Yu CH, Song GS, Yhee JY, et al. Histopathological and immunohistochemical comparison of the brain of human patients with Alzheimer’s disease and the brain of aged dogs with cognitive dysfunction. J Comp Pathol 2011;145:45-58. https://doi.org/10.1016/j.jcpa.2010.11.004
- Mutsuga M, Chambers JK, Uchida K, et al. Binding of curcumin to senile plaques and cerebral amyloid angiopathy in the aged brain of various animals and to neurofibrillary tangles in Alzheimer’s brain. J Vet Med Sci 2012;74:51-57. https://doi.org/10.1292/jvms.11-0307
- Finch CE, Austad SN. Commentary: is Alzheimer's disease uniquely human? Neurobiol Aging 2015 Feb;36:553-5. https://doi.org/10.1016/j.neurobiolaging.2014.10.025
- Languille S, Blanc S, Blin O, et al. The grey mouse lemur: a non-human primate model for ageing studies. Ageing Res Rev 2012;11:150-162. https://doi.org/10.1016/j.arr.2011.07.001
- Pérez SE, Raghanti MA, Hof PR, et al. Alzheimer's disease pathology in the neocortex and hippocampus of the weste.rn lowland gorilla (Gorilla gorilla gorilla). J Comp Neurol 2013;521:4318-4338. https://doi.org/10.1002/cne.23428
- Uchihara T, Endo K, Kondo H, et al. Tau pathology in aged cynomolgus monkeys is progressive supranuclear palsy/corticobasal degeneration, but not Alzheimer disease-like. Ultrastructural mapping of tau by EDX. Acta Neuropathol Commun 2016;4:118. https://doi.org/10.1186/s40478-016-0385-5
- Edler MK, Sherwood CC, Meindl RS, et al. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer's disease. Neurobiol Aging 2017;59:107-120. https://doi.org/10.1016/j.neurobiolaging.2017.07.006
- Takaichi Y, Chambers JK, Takahashi K, et al. Amyloid β and tau pathology in brains of aged pinniped species (sea lion, seal, and walrus). Amyloid β and tau pathology in brains of aged pinniped species (sea lion, seal, and walrus). Acta Neuropathol Commun 2021;9:10. https://doi.org/10.1186/s40478-020-01104-3
- Schultz C, Dehghani F, Hubbard GB, et al. Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J Neuropathol Exp Neurol 2000:59:39-52. https://doi.org/10.1093/jnen/59.1.39
- Schultz C, Hubbard GB, Rüb U, et al. Age-related progression of tau pathology in brains of baboons. Neurobiol Aging 2000;21: 905-912. https://doi.org/10.1016/s0197-4580(00)00176-7
- Abdel Rassoul R, Alves S, Pantesco V, et al. Distinct transcriptome expression of the temporal cortex of the primate Microcebus murinus during brain aging versus Alzheimer’s disease like pathology. PLoS ONE 2010;5: e12770. https://doi.org/10.1371/journal.pone.0012770
- Edler MK, Munger EL, Meindl RS, et al. Neuron loss associated with age but not Alzheimer's disease pathology in the chimpanzee brain. Philos Trans R Soc Lond B Biol Sci 2020;375:20190619. https://doi.org/10.1098/rstb.2019.0619
- Head E, Moffat K, Das P, et al. Beta-amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiol Aging 2005;26:749–763. https://doi.org/10.1016/j.neurobiolaging.2004.06.015
- Gunn-Moore DA, McVee J, Bradshaw JM, et al. Ageing changes in cat brains demonstrated by β-amyloid and AT8-immunoreactive phosphorylated tau deposits J Feline Med Surg 2006;8:234-242. https://doi.org/10.1016/j.jfms.2006.01.003
- Chambers JK, Tokuda T, Uchida K, et al. The domestic cat as a natural animal model of Alzheimer's disease. Acta Neuropathol Commun 2015;3:78. https://doi.org/10.118 /s40478-015-0258-3.
- Poncelet L, Ando K, Vergara C, et al. A 4R tauopathy develops without amyloid deposits in aged cat brains. Neurobiol Aging 2019;81:200e212. https://doi.org/10.1016/j.neurobiolaging.2019.05 024.
- Rapoport SI. Hypothesis: Alzheimer's disease is a phylogenetic disease. Med Hypotheses 1989; 29: 147-150. https://doi.org/10.1016/0306-9877(89)90185-0
- Gonatas NK, Anderson W, Evangelista I. The contribution of altered synapses in the senile plaques: an electron microscopic study in Alzheimer’s disease. J Neuropathol Exp Neurol 1967;26:25-39. https://doi.org/10.1097/00005072-196701000-00003
- Scheibel ME, Lindsay RD, Tomiyasu U, et al. Progressive dendritic changes in aging human cortex. Exp Neurol 1975;47:392-403. https://doi.org/10.1016/0014-4886(75)90072-2
- Buell SJ, Coleman PD. Dendritic growth in the aged human brain and failure of growth in senile dementia. Science 1979; 206:854-856. https://doi.org/10.1126/science.493989
- Buell SJ, Coleman PD. Quantitative evidence for selective dendritic growth in normal human aging but not in senile dementia. Brain Res 1981;214:23-41. https://doi.org/10.1016/0006-8993(81)90436-4
- Flood DG, Buell SJ, Defiore CH, et al. Age-related dendritic growth in dentate gyrus of human brain is followed by regression in the 'oldest old'. Brain Res 1985;345:366-368. https://doi.org/10.1016/0006-8993(85)91018-2
- Flood DG, Buell SJ, Horwitz GJ, et al. Dendritic extent in human dentate gyrus granule cells in normal aging and senile dementia. Brain Res 1987;402:205-216. https://doi.org/10.1016/0006-8993(87)90027-8
- Catalá I, Ferrer I, Galofré E, et al. Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative Golgi study on biopsy samples. Hum Neurobiol 1988;6:255-259. PMID: 3350705.
- Ferrer I, Guionnet N, Cruz-Sánchez F, et al. Neuronal alterations in patients with dementia: a Golgi study on biopsy samples. Neurosci Lett 1990;114:11-16. https://doi.org/10.1016/0304-3940(90)90420-e
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30: 572-580. https://doi.org/10.1002/ana.410300410
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002; 298:789-791. https://doi.org/10.1126/science.1074069
- Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging 2003;24:1029-1046. https://doi.org/10.1016/j.neurobiolaging.2003.08.002
- Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol 2009;118: 167-179. https://doi.org/10.1007/s00401-009-0536-x
- Domínguez-Álvaro M, Montero-Crespo M, Blazquez-Llorca L, et al. Three-dimensional analysis of synapses in the transentorhinal cortex of Alzheimer's disease patients. Acta Neuropathol Commun 2018;6:20. https://doi.org/10.1186/s40478-018-0520-6
- Montero-Crespo M, Domínguez-Álvaro M, Alonso-Nanclares L, et al. Three-dimensional analysis of synaptic organization in the hippocampal CA1 field in Alzheimer's disease. Brain 2021;144:553-573. https://doi.org/10.1093/brain/awaa406
- cheibel AB, Tomiyasu U. Dendritic sprouting in Alzheimer's presenile dementia. Exp Neurol 1978;60:1-8. https://doi.org/10.1016/0014-4886(78)90164-4
- Paula-Barbosa MM, Cardoso RM, Guimaraes ML, et al. Dendritic degeneration and regrowth in the cerebral cortex of patients with Alzheimer's disease. J Neurol Sci 1980;45:129-134. https://doi.org/10.1016/s0022-510x(80)80014-1
- Probst A, Basler V, Bron B, et al. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res 1983;268:249-254. https://doi.org/10.1016/0006-8993(83)90490-0
- Ferrer I, Aymami A, Rovira A, et al. Growth of abnormal neurites in atypical Alzheimer’s disease. A study with the Golgi method. Acta Neuropathol 1983;59:167-170. https://doi.org/10.1007/BF00703200
- Arendt T, Zvegintseva HG, Leontovich TA. Dendritic changes in the basal nucleus of Meynert and in the diagonal band nucleus in Alzheimer's disease--a quantitative Golgi investigation. Neuroscience 1986;19:1265-1278. https://doi.org/10.1016/0306-4522(86)90141-7
- Masliah E, Mallory M, Hansen L, et al. Patterns of aberrant sprouting in Alzheimer's disease. Neuron 1991;6:729-739. https://doi.org/10.1016/0896-6273(91)90170-5
- Masliah E, Mallory M, Hansen L, et al. Localization of amyloid precursor protein in GAP43-immunoreactive aberrant sprouting neurites in Alzheimer's disease. Brain Res 1992;574:312-316. https://doi.org/10.1016/0006-8993(92)90831-s
- Hashimoto M, Masliah E. Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer's and dementia with Lewy bodies. Neurochem Res 2003;28:1743-1756. https://doi.org/10.1023/a:1026073324672
- Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, et al. The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain 2013;136:1913-1928. https://doi.org/10.1093/brain/awt088
- Mijalkov M, Volpe G, Fernaud-Espinosa I, et al. Dendritic spines are lost in clusters in Alzheimer's disease. Sci Rep 2021;11:12350. https://doi.org/10.1038/s41598-021-91726-x
- Tsai J, Grutzendler J, Duff K, et al. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 2004;7:1181-1183. https://doi.org/10.1038/nn1335
- Garcia-Marin V, Blazquez-Llorca L, Rodriguez JR, et al. Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front Neuroanat 2009;3:28. https://doi.org/10.3389/neuro.05.028.2009
- Hoover BR, Reed MN, Su S, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010;68:1067-1081. https://doi.org/10.1016/j.neuron.2010.11.030
- Yin X, Zhao C, Qiu Y, et al. Dendritic/post-synaptic tau and early pathology of Alzheimer's disease. Front Mol Neurosci 2021;14:671779. https://doi.org/10.3389/fnmol.2021.671779
- Masliah E, Mallory M, Hansen L, et al. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 1994;174:67-72. https://doi.org/10.1016/0304-3940(94)90121-x
- Gouras GK, Willén K, Faideau M. The inside-out amyloid hypothesis and synapse pathology in Alzheimer's disease. Neurodegener Dis 2014;13:142-146. https://doi.org/10.1159/000354776
- Muntané G, Dalfó E, Martinez A, et al. Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer's disease, and in Parkinson's disease and related alpha-synucleinopathies. Neuroscience 2008;152:913-923. https://doi.org/10.1016/j.neuroscience.2008.01.030
- Gouras GK, Tampellini D, Takahashi RH, et al. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol 2010;119:523-541. https://doi.org/10.1007/s00401-010-0679-9
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 2010;13:812-818. https://doi.org/10.1038/nn.2583
- Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol 2012;4:a005777.
- Capetillo-Zarate E, Gracia L, Tampellini D, et al. Intraneuronal Aβ accumulation, amyloid plaques, and synapse pathology in Alzheimer's disease. Neurodegener Dis 2012;10:56-59. https://doi.org/10.1101/cshperspect.a005777
- Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 2014;82:756-771. https://doi.org/10.1016/j.neuron.2014.05.004
- Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer's disease neurons. J Alzheimers Dis 2017;57: 975-999. https://doi.org/10.3233/JAD-160612
- Li K, Wei Q, Liu FF, et al. Synaptic dysfunction in Alzheimer's disease: Abeta, Tau, and epigenetic alterations. Mol Neurobiol 2018;55:3021-3032. https://doi.org/10.1007/s12035-017-0533-3
- Chen Y, Fu AKY, Ip NY. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol Ther 2019;195:186-198. https://doi.org/10.1016/j.pharmthera.2018.11.006
- John A, Reddy PH. Synaptic basis of Alzheimer's disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res Rev 2021;65:101208. https://doi.org/10.1016/j.arr.2020.101208
- Coomans EM, Schoonhoven DN, Tuncel H, et al. In vivo tau pathology is associated with synaptic loss and altered synaptic function. Alzheimers Res Ther 2021;13:35. https://doi.org/10.1186/s13195-021-00772-0
- De Vos SL, Corjuc BT, Oakley DH, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci 2018;12: 267. https://doi.org/10.3389/fnins.2018.00267
- Pelucchi S, Stringhi R, Marcello E. Dendritic spines in Alzheimer's disease: How the actin cytoskeleton contributes to synaptic failure. Int J Mol Sci 2020;21:908. https://doi.org/10.3390/ijms21030908
- Kamat PK, Kalani A, Rai S, et al. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer's disease: Understanding the therapeutics strategies. Mol Neurobiol 2016;53:648-661. https://doi.org/10.1007/s12035-014-9053-6
- Arendt T, Brückner MK. Linking cell-cycle dysfunction in Alzheimer's disease to a failure of synaptic plasticity. Biochim Biophys Acta 2007;1772:413-421. https://doi.org/10.1016/j.bbadis.2006.12.005
- Li S, Sheng ZH. Energy matters: presynaptic metabolism and the maintenance of synaptic transmission. Nat Rev Neurosci 2022;23:4-22. https://doi.org/10.1038/s41583-021-00535-8
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 1976;2:1403. https://doi.org/10.1016/s0140-6736(76)91936-x
- Perry EK, Tomlinson BE, Blessed G, et al. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J 1978;2:1457-1459. https://doi.org/10.1136/bmj.2.6150.1457
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 1983;219:1184-1190. https://doi.org/10.1126/science.6338589
- Francis PT, Palmer AM, Snape M, et al. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 1999;66:137-147. https://doi.org/10.1136/jnnp.66.2.137
- Lees AJ, Tolosa E, Olanow CW. Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov Disord 2015;30:19-36. https://doi.org/10.1002/mds.26120
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int 2004;45:583-595. https://doi.org/10.1016/j.neuint.2004.03.007
- Parsons MP, Raimond LA. Extrasynaptic NMDA receptor Involvement in central nervous system disorders. Neuron 2014;82: 279-293. https://doi.org/10.1016/j.neuron.2014.03.030
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 1992;326:1464-1468. https://doi.org/10.1056/NEJM199205283262204
- Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 1990;11:379-387. https://doi.org/10.1016/0165-6147(90)90184-a
- Song J, Yanga X, Zhanga M, et al. Glutamate metabolism in mitochondria is closely related to Alzheimer’s disease. J Alzheimers Dis 2021;84:557-578. https://doi.org/10.3233/JAD-210595
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010; 11: 682-696. https://doi.org/10.1038/nrn2911
- Mufson EJ, Kordower JH. Cholinergic basal forebtrain system in the primate central nervous system: anatomy, connectivity, neurochemistry, aging, dementia, and experimental therapeutics. In: Functional neurobiology of aging. Hof PR, Mobbs CV (Eds.) Academic Press 2001, pp: 243-281
- Mishizen A, Ikonomovic M, Armstrong DM. Glutamate receptors in aging and Alzheimer’s disease. In: Functional neurobiology of aging. Hof PR, Mobbs CV (Eds.). Academic Press 2001, pp: 283-314
- Wang R, Reddy PH. Role of glutamate and NMDA receptors in alzheimer’s disease. J Alzheimers Dis 2017;57:1041-1048. https://doi.org/10.3233/JAD-160763
- Armada-Moreira A, Gomes JI, Pina CC, et al. Going the extra (Synaptic) mile: excitotoxicity as the road toward neurodegenerative diseases. Front Cell Neurosci 2020;14:90. https://doi.org/10.3389/fncel.2020.00090
- Šimić G, Babić Leko M, Wray S, et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog Neurobiol 2017;151:101-138. https://doi.org/10.1016/j.pneurobio.2016.04.001
- Sharma K, Pradhan S, Duffy L, et al. Role of receptors in relation to plaques and tanglers in Alzheimer’s disease pathology. Int J Mol Sci 2021;22:12987. https://doi.org/10.3390/ijms222312987
- Cai Z, Ratka A. Opioid system and Alzheimer's disease. Neuromolecular Med 2012;14:91-111. https://doi.org/10.1007/s12017-012-8180-3
- Morley JE. Peptides and aging: Their role in anorexia and memory. Peptides 2015;72:112-118. https://doi.org/10.1016/j.peptides 2015.04.007.
- Henry MS, Gendron L, Tremblay ME, et al. Enkephalins: endogenous analgesics with an emerging role in stress resilience. Neural Plast 2017;2017:1546125. https://doi.org/10.1155/2017/154612
- Valentino RJ, Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018;43:2514-2520. https://doi.org/10.1038/s41386-018-0225-3
- Garcia-Esparcia P, Schlüter A, Carmona M, et al. Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: novel putative chemoreceptors in the human brain. J Neuropathol Exp Neurol 2013;72:524-539. https://doi.org/10.1097/NEN.0b013e318294fd76
- Ferrer I, Garcia-Esparcia P, Carmona M, et al. Olfactory receptors in non-chemosensory organs: the nervous system in health and disease Front Aging Neurosci 2016; 8:163. https://doi.org/10.3389/fnagi.2016.00163
- Carlson AB, Kraus GP. Physiology, cholinergic receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526134
- Maelicke A. Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer’s disease. Dement Geriatr Cogn Disord 2000;11:11-18. https://doi.org/10.1159/000051227
- Cheng YJ, Lin CH, Lane HY. Involvement of cholinergic, adrenergic, and glutamatergic network modulation with cognitive dysfunction in Alzheimer's disease. Int J Mol Sci 2021; 22:2283. https://doi.org/10.3390/ijms22052283
- Mufson EJ, Bothwell M, Hersh LB, et al. Nerve growth factor receptor immunoreactive profiles in the normal, aged human basal forebrain: colocalization with cholinergic neurons. J Comp Neurol 1989;285:196-217. https://doi.org/10.1002/cne.902850204
- Mufson EJ, Bothwell M, Kordower JH. Loss of nerve growth factor receptor-containing neurons in Alzheimer's disease: a quantitative analysis across subregions of the basal forebrain. Exp Neurol 1989;105:221-232. https://doi.org/10.1016/0014-4886(89)90124-6
- Kordower JH, Gash DM, Bothwell M, et al. Nerve growth factor receptor and choline acetyltransferase remain colocalized in the nucleus basalis (Ch4) of Alzheimer's patients. Neurobiol Aging 1989;10:67-74. https://doi.org/10.1016/s0197-4580(89)80013-2
- Mufson EJ, Kordower EJ. Nerve growth factor in Alzheimer’s disease. In Cerebral cortex, Peters AA, Morrison JH, eds. Kluver Academic/Plenum, New York, 1999;14:681-731
- Whitehouse PJ, Price DL, Struble RG, et al. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982; 215:1237-1239. https://doi.org/10.1126/science.7058341
- DeKosky ST, Ikonomovic MD, Styren SD, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 2002;51:145-155. https://doi.org/10.1002/ana.10069
- Tiernan CT, Ginsberg SD, He B, et al. Pretangle pathology within cholinergic nucleus basalis neurons coincides with neurotrophic and neurotransmitter receptor gene dysregulation during the progression of Alzheimer's disease. Neurobiol Dis 2018;117:125-136. https://doi.org/10.1016/j.nbd.2018.05.021
- Ferreira-Vieira TH, Guimaraes IM, Silva FR, et al. Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol 2016;14:101-115. https://doi.org/10.2174/1570159x13666150716165726
- Perry EK, Morris CM, Court JA, et al. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience 1995;64:385-395. https://doi.org/10.1016/0306-4522(94)00410-7
- Court J, Martin-Ruiz C, Piggott M, et al. Nicotinic receptor abnormalities in Alzheimer’s disease. Biol Psychiatry 2001;49:175-184. https://doi.org/10.1016/s0006-3223(00)01116-1
- Pimlott SL, Piggott M, Owens J, et al. Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[125I]-A-85380. Neuropsychopharmacology 2004;29:108-116. https://doi.org/10.1038/sj.npp.1300302
- Shen J, Wu J. Nicotinic cholinergic mechanisms in Alzheimer’s disease. Int Rev Neurobiol 2015;124:275-292. https://doi.org/10.1016/bs.irn.2015.08.002
- Ono K, Hasegawa K, Yamada M, et al. Nicotine breaks down preformed Alzheimer’s β-amyloid fibrils in vitro. Biol Psychiatry 2002; 52:880-886. https://doi.org/10.1016/s0006-3223(02)01417-8
- Wang HY, Lee DH, D’Andrea MR, et al. β-amyloid1-42 binds to α7 nicotinic acetylcholine receptor with high affinity: Implications for Alzheimer’s disease pathology. J Biol Chem 2000; 275: 5626-5632. https://doi.org/10.1074/jbc.275.8.5626
- Liu Q, Xie X, Lukas RJ, et al. A novel nicotinic mechanism underlies beta-amyloidinduced neuronal hyperexcitation. J Neurosci 2013;33:7253-7263. https://doi.org/10.1523/JNEUROSCI.3235-12.2013
- Liu Q, Xie X, Emadi S, et al. A novel nicotinic mechanism underlies beta-amyloid induced neurotoxicity. Neuropharmacology 2015;97:457-463. https://doi.org/10.1016/j.neuropharm.2015.04.025
- Wang HY, Li W, Benedetti NJ, et al. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem 2003; 278:31547-31553. https://doi.org/10.1074/jbc.M212532200
- Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 2004; 89:337-343. https://doi.org/10.1046/j.1471-4159.2004.02347.x
- Sadigh-Eteghad S, Majdi A, Mahmoudi J, et al. Astrocytic and microglial nicotinic acetylcholine receptors: an overlooked issue in Alzheimer's disease. J Neural Transm 2016;1231359-1367. https://doi.org/10.1007/s00702-016-1580-z
- Lebois EP, Thorn C, Edgerton JR, et al. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018; 136: 362-373. https://doi.org/10.1016/j.neuropharm.2017.11.018
- Volpicelli LA, Allan IL. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog Brain Res 2004;145:59-66. https://doi.org/10.1016/S0079-6123(03)45003-6
- Ishii M, Kurachi Y. Muscarinic acetylcholine receptors. Curr Pharm Des 2006;12: 3573-3581. https://doi.org/10.2174/138161206778522056
- Kudlak M, Tadi P. Physiology, Muscarinic Receptor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Pavía J, de Ceballos ML, Sanchez de la Cuesta F. Alzheimer's disease: relationship between muscarinic cholinergic receptors, beta-amyloid and tau proteins. Fundam Clin Pharmacol 1998;12:473-481. https://doi.org/10.1111/j.1472-8206.1998.tb00975.x
- Clader JW, Wang Y. Muscarinic receptor agonists and antagonists in the treatment of Alzheimer’s disease. Curr Pharm Des 2005;11:3353-3361. https://doi.org/10.2174/138161205774370762
- Fisher A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008; 5:433-442. https://doi.org/10.1016/j.nurt.2008.05.002
- Davis AA, Fritz JJ, Wess J, et al. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci 2010;30:4190-4196. https://doi.org/10.1523/JNEUROSCI.6393-09.2010
- Geula C, Mesulam M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res 1989;498: 185-189. https://doi.org/10.1016/0006-8993(89)90419-8
- Sberna G, Sáez-Valero J, Beyreuther K, et al. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J Neurochem 1997;69:1177-1184. https://doi.org/10.1046/j.1471-4159.1997.69031177.x
- Furukawa H, Singh SK, Mancusso R, et al. Subunit arrangement and function in NMDA receptors. Nature 2005;438:185-192. https://doi.org/10.1038/nature04089
- Yamamoto H, Hagino Y, Kasai S, et al. Specific roles of NMDA receptor subunits in mental disorders. Curr Mol Med 2015; 15: 193-205. https://doi.org/10.2174/1566524015666150330142807
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nature Rev Neurosci 2008; 9:423. https://doi.org/10.1038/nrn2379
- Steinhäuser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci 1996;19: 339-345. https://doi.org/10.1016/0166-2236(96)10043-6
- Shemer I, Holmgren C, Min R, et al. Non-fibrillar beta-amyloid abates spike-timing-dependent synaptic potentiation at excitatory synapses in layer 2/3 of the neocortex by targeting postsynaptic AMPA receptors. Eur J Neurosci 2006;23:2035-47. https://doi.org/10.1111/j.1460-9568.2006.04733.x
- Parameshwaran K, Dhanasekaran M, Suppiramaniam V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol 2008;210:7-13. https://doi.org/10.1016/j.expneurol.2007.10.008
- Ferreira IL, Bajouco LM, Mota SI, et al. Amyloid beta peptide 1-42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012;51:95-106. https://doi.org/10.1016/j.ceca.2011.11.008
- Decker H, Jürgensen S, Adrover MF, et al. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-β peptide oligomers. J Neurochem 2010;115:1520-1529. https://doi.org/10.1111/j.1471-4159.2010.07058.x Epub 2010 Nov 11.
- Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Abeta production in Alzheimer's disease. Rev Neurosci 2011;22:285-294. https://doi.org/10.1515/RNS.2011.029
- Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in Alzheimer's disease - A focus on NMDA receptors. Neuropharmacology 2014;76:16-26. https://doi.org/10.1016/j.neuropharm.2013.08.013
- Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer's disease. Neurol Sci 2016;37:1039-1047. https://doi.org/10.1007/s10072-016-2546-5
- Foster TC, Kyritsopoulos C, Kumar A. Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer's disease. Behav Brain Res 2017;322:223-232. https://doi.org/10.1016/j.bbr.2016.05.012
- Babaei P. NMDA and AMPA receptors dysregulation in Alzheimer's disease. Eur J Pharmacol 2021;908:174310. https://doi.org/10.1016/j.ejphar.2021.174310
- Reinders NR, Pao Y, Renner MC, et al. Effects of amyloid-β require AMPAR subunit GluA3. Proc Natl Acad Sci USA 2016;113: E6526-E6534. https://doi.org/10.1073/pnas.1614249113
- Zhang Y, Guo O, Huo Y, et al. Amyloid-β induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer’s disease. J Alzheimer’s Dis 2018;62:1789-1801. https://doi.org/10.3233/JAD-170879
- Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1-105. https://doi.org/10.1016/s0301-0082(00)00067-8
- Kirvell SL, Esiri M, Francis PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem 2006;98:939-950. https://doi.org/10.1111/j.1471-4159.2006.03935.x
- Jacob CP, Koutsilieri E, Bartl J, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimers Dis 2007; 11:97-116. https://doi.org/10.3233/jad-2007-11113
- Pow DV, Cook DG.Neuronal expression of splice variants of "glial" glutamate transporters in brains afflicted by Alzheimer's disease: unmasking an intrinsic neuronal property. Neurochem Res 2009;34:1748-1757. https://doi.org/10.1007/s11064-009-9957-0
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 1997; 37: 205-237. https://doi.org/10.1146/annurev.pharmtox.37.1.205
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology 1995;3:1-26. https://doi.org/10.1016/0028-3908(94)00129-g
- Crupi R, Impellizeri D, Cuzzocrea S. Role of metabotropic glutamate receptors in neurological disorders. Front Mol Neurosci 2019;12:20. https://doi.org/10.3389/fnmol.2019.00020
- Mao LM, Bodepudi A, Chu XP, et al. Group I metabotropic glutamate receptors and interacting partners: an update. Int J Mol Sci 2022;23:840. https://doi.org/10.3390/ijms23020840
- Albasanz JL, Dalfó E, Ferrer I, et al. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer's disease and dementia with Lewy bodies correlates with stage of Alzheimer's-disease-related changes. Neurobiol Dis 2005;20:685-693. https://doi.org/10.1016/j.nbd.2005.05.001
- Goetz T, Arslan A, Wisden W, et al. GABA(A) receptors: Structure and function in the basal ganglia. Prog Brain Res 2007;160:21-41. https://doi.org/10.1016/S0079-6123(06)60003-4
- Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol Sci 2010;58:123-147. https://doi.org/10.1016/S1054-3589(10)58006-2
- Jembrek MJ, Vlainic J. GABA receptors: pharmacological potential and pitfalls. Curr Pharm Des 2015;21:4943-4959. https://doi.org/10.2174/1381612821666150914121624
- Terunuma M. Diversity of structure and function of GABA(B) receptors: a complexity of GABA(B)-mediated signaling. Proc Jpn Acad Ser B Phys Biol Sci 2018;94:390-411. https://doi.org/10.2183/pjab.94.026
- Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641-653. https://doi.org/10.1111/j.1471-4159.2006.03913.x
- Leke R, Schousboe A. The glutamine transporters and their role in the glutamate/GABA-glutamine cycle. Adv Neurobiol 2016;13:223-257. https://doi.org/10.1007/978-3-319-45096-4_8
- Yoon BE, Lee CJ. GABA as a rising gliotransmitter. Front Neural Circuits 2014;8:141. https://doi.org/10.3389/fncir.2014.00141
- Ambrad Giovannetti E, Fuhrmann M. Unsupervised excitation: GABAergic dysfunctions in Alzheimer's disease. Brain Res 2019;1707:216-226. https://doi.org/10.1016/j.brainres.2018.11.042
- Calvo-Flores Guzmán B, Vinnakota C, Govindpani K, et al. The GABAergic system as a therapeutic target for Alzheimer's disease. J Neurochem 2018;146:649-669. https://doi.org/10.1111/jnc.14345
- Govindpani K, Calvo-Flores Guzmán B, et al. Towards a better understanding of GABAergic remodeling in Alzheimer's disease. Int J Mol Sci 2017;18: 1813. https://doi.org/10.3390/ijms18081813
- Riese F, Gietl A, Zölch N, et al. Posterior cingulate γ-aminobutyric acid and glutamate/glutamine are reduced in amnestic mild cognitive impairment and are unrelated to amyloid deposition and apolipoprotein E genotype. Neurobiol Aging 2015;36:53-59. https://doi.org/10.1016/j.neurobiolaging.2014.07.030
- Huang D, Liu D, Yin J, et al. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur Radiol 2017;27:2698-2705. https://doi.org/10.1007/s00330-016-4669-8
- Murari G, Liang DR, Ali A, et al. Prefrontal GABA Levels Correlate with Memory in Older Adults at High Risk for Alzheimer's Disease. Cereb Cortex Commun 2020;1:tgaa022. https://doi.org/10.1093/texcom/tgaa02
- Liu H, Zhang D, Lin H, et al. Meta-analysis of neurochemical changes estimated via magnetic resonance spectroscopy in mild cognitive impairment and Alzheimer's disease. Front Aging Neurosci 2021;13:738971. https://doi.org/10.3389/fnagi.2021.738971
- Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci USA 2012;109:10071-10076. https://doi.org/10.1073/pnas.1204606109
- Mizukami K, Ikonomovic MD, Grayson DR, et al. Immunohistochemical study of GABAA receptor alpha1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res 1998;799:148-155. https://doi.org/10.1016/s0006-8993(98)00437-5
- Rissman RA, Mishizen-Eberz AJ, Carter TL, et al. Biochemical analysis of GABA(A) receptor subunits alpha 1, alpha 5, beta 1, beta 2 in the hippocampus of patients with Alzheimer's disease neuropathology. Neuroscience 2003;120:695-704. https://doi.org/10.1016/s0306-4522(03)00030-7
- Rissman RA, Bennett DA, Armstrong DM. Subregional analysis of GABA(A) receptor subunit mRNAs in the hippocampus of older persons with and without cognitive impairment. J Chem Neuroanat 2004;28:17-25. https://doi.org/10.1016/j.jchemneu.2004.05.003
- Iwakiri M, Mizukami K, Ikonomovic DM, et al. An immunohistochemical study of GABA A receptor gammasubunits in Alzheimer's disease hippocampus: relationship to neurofibrillary tangle progression. Neuropathology 2009;29:263-269. https://doi.org/10.1111/j.1440-1789.2008.00978.x
- Mizukami K, Ikonomovic MD, Grayson DR, et al. Immunohistochemical study of GABA(A) receptor beta2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp Neurol 1997;147:333-345. https://doi.org/10.1006/exnr.1997.6591
- Kwakowsky A, Calvo-Flores Guzmán B, Pandya M, et al. GABAA receptor subunit expression changes in the human Alzheimer's disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J Neurochem 2018;145:374-392. https://doi.org/10.1111/jnc.14325
- Villette V, Poindessous-Jaza F, Simon A, et al. Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-beta pathology in the rat. J Neurosci 2010;30:10991-11003. https://doi.org/10.1523/JNEUROSCI.6284-09.2010
- Verret L, Mann EO, Hang GB, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012;149:708-721. https://doi.org/10.1016/j.cell.2012.02.046
- Ulrich D. Amyloid-beta impairs synaptic inhibition via GABA(A) receptor endocytosis. J Neurosci 2015;35: 9205-9210. https://doi.org/10.1523/JNEUROSCI.0950-15.2015
- Hamm V, Héraud C, Bott JB, et al. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer's disease. Sci Adv 2017;3:e1601068. https://doi.org/10.1126/sciadv.1601068
- Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodelling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 2007;55:697-711. https://doi.org/10.1016/j.neuron.2007.07.025
- Hoyer D, Martin G. 5-HT receptor classification and nomenclature: Towards a harmonization with the human genome. Neuropharmacology 1997;36:419-428. https://doi.org/10.1016/s0028-3908(97)00036-1
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology 1999;38:1083-1152. https:/ doi.org/10.1016/s0028-3908(99)00010-6.
- Leysen JE. 5-HT2 receptors. Curr Drug Targets-CNS Neurol Disord 2004;3:11-26. https://doi.org/10.2174/1568007043482598
- Fink KB, Göthert M. 5-HT receptor regulation of neurotransmitter release. Pharmacol Rev 2007;59:360-417. https://doi.org/10.1124/pr.107.07103
- Nichols DE, Nichols CD. Serotonin receptors. Chem Rev 2008;108:1614-1641. https://doi.org/10.1021/cr078224o
- Barnes NM, Hales TG, Lummis SC, Peters JA. The 5-HT3 receptor: the relationship between structure and function. Neuropharmacology 2009;56:273-284. https://doi.org/10.1016/j.neuropharm.2008.08.003
- Berumen LC, Rodríguez A, Miledi R, et al. Serotonin receptors in hippocampus. Sci World J 2012; 2012:823493. https://doi.org/10.1100/2012/823493
- Palacios JM. Serotonin receptors in brain revisited. Brain Res 2016;1645:46-49. https://doi.org/10.1016/j.brainres.2015.12.042
- Wirth A, Holst K, Ponimaskin E. How serotonin receptors regulate morphogenic signalling in neurons. Prog Neurobiol 2017;151:35-56. https://doi.org/10.1016/j.pneurobio.2016.03.007
- Consolo S, Arnaboldi S, Giorgi S, et al. 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. Neuroreport 1994;5:1230-1232. https://doi.org/10.1097/00001756-199406020-00018
- Woods S, Clarke NN, Layfield R, et al. 5-HT (6) receptor agonists and antagonists enhance learning and memory in a conditioned emotion response paradigm by modulation of cholinergic and glutamatergic mechanisms. Br J Pharmacol 2012;167: 436-449. https://doi.org/10.1111/j.1476-5381.2012.02022.x
- Seyedabadi M, Fakhfouri G, Ramezani V, et al. The role of serotonin in memory: interactions with neurotransmitters and downstream signaling. Exp Brain Res 2014;232:723-738. https://doi.org/10.1007/s00221-013-3818-4
- De Deurwaerdere P, Di Giovanni G. 5-HT interaction with other neurotransmitters: An overview. Prog Brain Res 2021;259:1-5. https://doi.org/10.1016/bs.pbr.2021.01.001
- Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci 2002;22:9453-9464. https://doi.org/10.1523/JNEUROSCI.22-21-09453.2002
- Noristani HN, Olabarria M, Verkhratsky A, et al. Serotonin fibre sprouting and increase in serotonin transporter immunoreactivity in the CA1 area of hippocampus in a triple transgenic mouse model of Alzheimer's disease. Eur J Neurosci 2010;32:71-79. https://doi.org/10.1111/j.1460-9568.2010.07274.x
- Rodríguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer's disease. Prog Neurobiol 2012;99:15-41. https://doi.org/10.1016/j.pneurobio.2012.06.010
- Švob Štrac D, Pivac N, Mück-Šeler D. The serotoninergic system and cognitive function. Transl Neurosci 2016;7:35-49. https://doi.org/10.1515/tnsci-2016-0007
- Meltzer CC, Smith G, DeKosky ST, et al. Serotonin in aging, late-life depression, and Alzheimer’s disease: the emerging role of functional imaging. Neuropsychopharmacology 1998;18:407-430. https://doi.org/10.1016/S0893-133X(97)00194-2
- Michelsen KA, Prickaerts J, Steinbusch HW. The dorsal raphe nucleus and serotonin: implications for neuroplasticity linked to major depression in Alzheimer’s disease. Prog Brain Res 2008;172:233-264. https://doi.org/10.1016/S0079-6123(08)00912-6
- Yun HM, Park KR, Kim EC, et al. Serotonin 6 receptor controls Alzheimer's disease and depression. Oncotarget 2015;6:26716-26728. https://doi.org/10.18632/oncotarget.5777
- Burke AD, Goldfarb D, Bollam P, et al. Diagnosing and treating depression in patients with Alzheimer's disease. Neurol Ther 2019;8:325-350. https://doi.org/10.1007/s40120-019-00148-5
- Cachard-Chastel M, Lezoualc’h F, Dewachter I, et al. 5-HT4 receptor agonists increase sAPPalpha levels in the cortex and hippocampus of male C57BL/6j mice. Br J Pharmacol 2007;150:883-892. https://doi.org/10.1038/sj.bjp.0707178
- Cirrito JR, Disabato BM, Restivo JL, et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc Natl Acad Sci USA 2011;108:14968-14973. https://doi.org/10.1073/pnas.1107411108
- Cochet M, Donneger R, Cassier E, et al. 5-HT4 receptors constitutively promote the non-amyloidogenic pathway of APP cleavage and interact with ADAM10. ACS Chem Neurosci 2013;4: 130-140. https://doi.org/10.1021/cn300095t
- Giannoni P, Gaven F, de Bundel D, et al. Early administration of RS 67333, a specific 5-HT4 receptor agonist, prevents amyloidogenesis and behavioral deficits in the 5XFAD mouse model of Alzheimer’s disease. Front Aging Neurosci 2013;5:96. https://doi.org/10.3389/fnagi.2013.00096
- Moser PC, Bergis OE, Jegham S, et al. SL65.0155, a novel 5-hydroxytryptamine(4) receptor partial agonist with potent cognition-enhancing properties. J Pharmacol Exp Ther 2002;302:731-741. https://doi.org/10.1124/jpet.102.034249
- Lezoualc'h F. 5-HT4 receptor and Alzheimer's disease: the amyloid connection. Exp Neurol 2007;205:325-329. https://doi.org/10.1016/j.expneurol.2007.02.001
- Cho S, Hu Y. Activation of 5-HT4 receptors inhibits secretion of beta-amyloid peptides and increases neuronal survival. Exp Neurol 2007;203:274-278. https://doi.org/10.1016/j.expneurol.2006.07.021
- Geldenhuys WJ, Van der Schyf CJ. Role of serotonin in Alzheimer’s disease: a new therapeutic target? CNS Drugs 2011;25:765-781. https://doi.org/10.2165/11590190-000000000-00000
- Werner FM, Covenas R. Serotonergic drugs: agonists/antagonists at specific serotonergic subreceptors for the treatment of cognitive, depressant and psychotic symptoms in Alzheimer's disease. Curr Pharm Des 2016;22:2064-2071. https://doi.org/10.2174/1381612822666160127113524
- Magierski R, Sobow T. Serotonergic drugs for the treatment of neuropsychiatric symptoms in dementia. Expert Rev Neurother 2016;16:375-387. https://doi.org/10.1586/14737175.2016.1155453
- Ferrero H, Solas M, Francis PT, et al. Serotonin 5-HT(6) receptor antagonists in Alzheimer's disease: therapeutic rationale and current development status. CNS Drugs 2017;31:19-32. https://doi.org/10.1007/s40263-016-0399-3
- Brendel M, Sauerbeck J, Greven S, et al. Serotonin selective reuptake inhibitor treatment improves cognition and grey matter atrophy but not amyloid burden during two-year follow-up in mild cognitive impairment and Alzheimer's disease patients with depressive symptoms. J Alzheimers Dis 2018;65:793-806. https://doi.org/10.3233/JAD-170387
- Manzoor S, Hoda N. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer's disease agents: A review. Eur J Med Chem 2020;206:112787. https://doi.org/10.1016/j.ejmech.2020 112787.
- Mdawar B, Ghossoub E, Khoury R. Selective serotonin reuptake inhibitors and Alzheimer’s disease. Regen Res 2020;15:41-46. https://doi.org/10.4103/1673-5374.264445
- Kucwaj-Brysz K, Baltrukevich H, Czarnota K, et al. Chemical update on the potential for serotonin 5-HT(6) and 5-HT(7) receptor agents in the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2021;49:128275. https://doi.org/10.1016/j.bmcl.2021.128275
- Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature 1991;350:350-354. https://doi.org/10.1038/350350a0
- Eiden LE, Schäfer MK, Weihe E, et al. The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflügers Arch. 2004;447: 636-640. https://doi.org/10.1007/s00424-003-1100-5
- Ruffolo RR, Hieble JP. Alpha-adrenoreceptors. Pharmacol Ther 1994;61:1-64. https://doi.org/10.1016/0163-7258(94)90058-2
- Musacchio JM. Enzymes involved in the biosynthesis and degradation of catecholamines. In: Biochemistry of Biogenic Amines. Iverson L (ed). Springer 2013, pp:1-35.
- Pickel VM. Extrasynaptic distribution of monoamine transporters and receptors. Prog Brain Res 2000;125:267-276. https://doi.org/10.1016/S0079-6123(00)25016-4
- Perez DM. α1-Adrenergic receptors in neurotransmission, synaptic plasticity, and cognition. Front Pharmacol 2020;11:581098. https://doi.org/10.3389/fphar.2020.581098
- Moore RY, Bloom FE. Central catecholamine neuron systems: Anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci 1979; 2: 113-168. https://doi.org/10.1146/annurev.ne.02.030179.000553
- Szabadi E. Functional neuroanatomy of the central noradrenergic system. J Psychopharmacol 2013;27:659-693. https://doi.org/10.1177/0269881113490326
- Poe GR, Foote S, Eschenko O, et al. Locus coeruleus: A new look at the blue spot. Nat Rev Neurosci 2020;21:644-659. https://doi.org/10.1038/s41583-020-0360-9
- Starke K. Presynaptic autoreceptors in the third decade: Focus on alpha2-adrenoceptors. J Neurochem 2001;78: 685-693. https://doi.org/10.1046/j.1471-4159.2001.00484.x
- Nicholas AP, Pieribone V, Hökfelt T. Distributions of mRNAs for alpha-2 adrenergic receptor subtypes in rat brain: an in situ hybridization study. J Comp Neurol 1993;328: 575-594. https://doi.org/10.1002/cne.903280409
- Day HE, Campeau S, Watson SJ Jr, et al. Distribution of alpha 1a-, alpha 1b- and alpha 1d-adrenergic receptor mRNA in the rat brain and spinal cord. J Chem Neuroanat 1997;13:115-139. https://doi.org/10.1016/s0891-0618(97)00042-2
- Pieribone VA, Nicholas AP, Dagerlind A, et al. Distribution of alpha 1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci 1994;14:4252-4268. https://doi.org/10.1523/JNEUROSCI.14-07-04252.1994
- Domyancic AV, Morilak DA. Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J Comp Neurol 1997;386:358-378. https://doi.org/10.1002/(sici)1096-9861(19970929)386:3<358::aid-cne3>3.0.co;2-0
- Benarroch EE. Locus coeruleus. Cell Tissue Res 2018;373:221-232. https://doi.org/10.1007/s00441-017-2649-1
- Paspalas CD Papadopoulos GC. Ultrastructural relationships between noradrenergic nerve fibers and non-neuronal elements in the rat cerebral cortex. Glia 1996;17:133-146. https://doi.org/10.1002 (SICI)1098-1136(199606)17:2<133::AID-GLIA5>3.0.CO;2-3.
- Yoshioka Y, Negoro R, Kadoi H, et al. Noradrenaline protects neurons against H2O2-induced death by increasing the supply of glutathione from astrocytes via beta3-adrenoceptor stimulation. J Neurosci Res. 2021 Feb;99(2):621-637. https://doi.org/10.1002/jnr.24733
- Giorgi FS, Galgani A, Puglisi-Allegra S, et al., Locus coeruleus and neurovascular unit: From its role in physiology to its potential role in Alzheimer’s disease pathogenesis. J Neurosci Res 2020;98:2406-2434. https://doi.org/10.1002/jnr.24718
- Feinstein DL, Heneka MT, Gavrilyuk V, et al. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int 2002;41:357-365. https://doi.org/10.1016/s0197-0186(02)00049-9
- Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab 2012;32:2135-2145. https://doi.org/10.1038/jcbfm.2012.115
- Wirth KJ. Role of noradrenergic brain nuclei in the regulation of carotid artery blood flow: pharmacological evidence from anesthetized pigs with alpha-2 adrenergic receptor modulator drugs. J Alzheimers Dis 2018;66:407-419. https://doi.org/10.3233/JAD-180340
- Descarries L, Mechawar N. Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system. Prog Brain Res 2000;125:27-47. https://doi.org/10.1016/S0079-6123(00)25005-X
- Watabe AM, Zaki PA, O'Dell TJ. Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci 2000;20: 5924-5931. https://doi.org/10.1523/JNEUROSCI.20-16-05924.2000
- Giorgi FS, Biagioni F, Galgani A, et al. Locus Coeruleus modulates neuroinflammation in parkinsonism and dementia. Int J Mol Sci 2020; 21: 8630. https://doi.org/10.3390/ijms21228630
- Brown RE, Sergeeva OA, Eriksson KS, et al. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci 2002;22: 8850-8859. https://doi.org/10.1523/JNEUROSCI.22-20-08850.2002
- Morilak DA, Barrera G, Echevarria DJ, et al. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry 2005;29:1214-1224. https://doi.org/10.1016/j.pnpbp.2005.08.007
- Zarow C, Lyness SA, Mortimer JA, et al. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 2003; 60:337-341. https://doi.org/10.1001/archneur.60.3.337
- Mravec B, Lejavova K, Cubinkova V. Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 2014; 11: 992-1001. https://doi.org/10.2174/1567205011666141107130505
- Ross JA, McGonigle P, Van Bockstaele EJ. Locus coeruleus, norepinephrine and Aβ peptides in Alzheimer's disease. Neurobiol Stress 2015;2:73-84. https://doi.org/10.1016/j.ynstr.2015.09.002
- Bekdash RA. The cholinergic system, the adrenergic system and the neuropathology of Alzheimer's disease. Int J Mol Sci 2021;22:1273. https://doi.org/10.3390/ijms22031273
- Lamerand S, Shahidehpour R, Ayala I, et al. Calbindin-D28K, parvalbumin, and calretinin in young and aged human locus coeruleus. Neurobiol Aging 2020;94:243-249. https://doi.org/10.1016/j.neurobiolaging.2020.06.006
- Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part II: Physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol 2008;6:254-285. https://doi.org/10.2174/157015908785777193
- Trillo L, Das D, Hsieh W, et al. Ascending monoaminergic systems alterations in Alzheimer's disease: translating basic science into clinical care. Neurosci Biobehav Rev 2013;37:1363-1679. https://doi.org/10.1016/j.neubiorev.2013.05.008
- Patthy Á, Murai J, Hanics J, et al. Neuropathology of the brainstem to mechanistically understand and to treat Alzheimer's disease. J Clin Med 2021;10:1555.
- Cross AJ, Crow TJ, Perry EK, et al. Reduced dopamine beta-hydroxylase activity in Alzheimer's disease. Br Med J 1981;282:93-94. https://doi.org/10.1136/bmj.282.6258.93
- Perry EK, Tomlinson BE, Blessed G, et al. Neuropathological and biochemical observations on the noradrenergic system in Alzheimer's disease. J Neurol Sci 1981;51:279-287. https://doi.org/10.1016/0022-510x(81)90106-4
- Mustapić M, Presečki P, Pivac N, et al. Genotype-independent decrease in plasma dopamine β-hydroxylase activity in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry 2013;44:94-99. https://doi.org/10.1016/j.pnpbp.2013.02.002
- Serra L, D’Amelio M, Di Domenico C, et al. In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer’s disease. Neurobiol Aging 2018;72:72-82. https://doi.org/10.1016/j.neurobiolaging.2018.08.012
- Schöne C, Burdakov D. Orexin/hypocretin and organizing principles for a diversity of wake-promoting neurons in the brain. Curr Top Behav Neurosci 2017;33:51-74. https://doi.org/10.1007/7854_2016_45
- Sakurai T, Saito YC, Yanagisawa M. Interaction between orexin neurons and monoaminergic systems. Front Neurol Neurosci 2021;45:11-21. https://doi.org/10.1159/000514955
- Liguori C. Orexin and Alzheimer's disease. Curr Top Behav Neurosci 2017;33:305-322. https://doi.org/10.1007/7854_2016_50
- Um YH, Lim HK. Orexin and Alzheimer's disease: A new perspective. Psychiatry Investig 2020;17:621-626. https://doi.org/10.30773/pi.2020.0136
- Dauvilliers Y. Hypocretin/orexin, sleep and Alzheimer's disease. Front Neurol Neurosci 2021;45:139-149. https://doi.org/10.1159/000514967
- Fredholm BB, IJzerman AP, Jacobson KA, et al. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 2001;53:527-552. PMID: 11734617
- Fredholm BB, IJzerman AP, Jacobson KA, et al. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors: An update. Pharmacol Rev 2011;63:1-34. https://doi.org/10.1124/pr.110.003285
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem Int 2001;38:107-125. https://doi.org/10.1016/s0197-0186(00)00034-6
- Fredholm BB, Chen JF, Cunha RA, et al. Adenosine and brain function. Int Rev Neurobiol 2005;63:191-270. https://doi.org/10.1016/S0074-7742(05)63007-3
- Sheth S, Brito R, Mukherjea D, et al. Adenosine receptors: expression, function and regulation. Int J Mol Sci 2014; 15: 2024-2052. https://doi.org/10.3390/ijms15022024
- Sebastiao AM, Ribeiro JA. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog Neurobiol 1996;48:167-189. https://doi.org/10.1016/0301-0082(95)00035-6
- Cunha RA, Almeida T, Ribeiro JA. Modification by arachidonic acid of extracellular adenosine metabolism and neuromodulatory action in the rat hippocampus. J Biol Chem 2000;275:37572-37581. https://doi.org/10.1074/jbc.M003011200
- Brand A, Vissiennon Z, Eschke D, et al. Adenosine A(1) and A(3) receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 2001;40:85–95. https://doi.org/10.1016/s0028-3908(00)00117-9
- Hettinger BD, Lee A, Linden J, et al. Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J Comp Neurol 2001;431:331-346. https://doi.org/10.1002/1096-9861(20010312)431:3<331::aid-cne1074>3.0.co;2-w
- Lopes LV, Cunha RA, Kull B, et al. Adenosine A(2A) receptor facilitation of hippocampal synaptic transmission is dependent on tonic A(1) receptor inhibition. Neuroscience 2002;112:319-329. https://doi.org/10.1016/s0306-4522(02)00080-5
- Rebola N, Oliveira CR, Cunha RA. Transducing system operated by adenosine A(2A) receptors to facilitate acetylcholine release in the rat hippocampus. Eur J Pharmacol 2002;454:31-38. https://doi.org/10.1016/s0014-2999(02)02475-5
- Rebola N, Rodrigues RJ, Oliveira CR, et al. Different roles of adenosine A1, A2A and A3 receptors in controlling kainate-induced toxicity in cortical cultured neurons. Neurochem Int 2005;47:317-325. https://doi.org/10.1016/j.neuint.2005.05.009
- Kalaria RN, Sromek S,Wilcox BJ, et al. Hippocampal adenosine A1 receptors are decreased in Alzheimer’s disease. Neurosci Lett 1990;118:257-260. https://doi.org/10.1016/0304-3940(90)90641-l
- Ulas J, Brunner LC, Nguyen L, et al. Reduced density of adenosine A1 receptors and preserved coupling of adenosine A1 receptors to G proteins in Alzheimer hippocampus: a quantitative autoradiographic study. Neuroscience 1993;52:843-854. https://doi.org/10.1016/0306-4522(93)90533-l
- Streit WJ, Braak H, Xue QS, et al. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475-485. https://doi.org/10.1007/s00401-009-0556-6
- Deckert J, Abel F, Kunig G, et al. Loss of human hippocampal adenosine A1 receptors in dementia: evidence for lack of specificity. Neurosci Lett 1998;244:1-4. https://doi.org/10.1016/s0304-3940(98)00108-6
- Angulo E, Casado V, Mallol J, et al. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol 2003;13:440-451. https://doi.org/10.1111/j.1750-3639.2003.tb00475.x
- Albasanz JL, Perez S, Barrachina M, et al. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease brain pathology Brain Pathol 2008;18:211-219. https://doi.org/10.1111/j.1750-3639.2007.00112
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat 2002;68-69: 619-631. https://doi.org/10.1016/s0090-6980(02)00060-6
- Devane WA, Axelrod J. Enzymatic synthesis of anandamide, an endogenous ligand for the cannabinoid receptor, by brain membranes. Proc Natl Acad Sci USA 1994;91:6698-6701. https://doi.org/10.1073/pnas.91.14.6698
- Sugiura T, Waku K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem Phys Lipids 2000;108:89-106. https://doi.org/10.1016/s0009-3084(00)00189-4
- Sugiura T, Waku K. Cannabinoid receptors and their endogenous ligands. J Biochem 2002; 132:7-12. https://doi.org/10.1093/oxfordjournals.jbchem.a003200
- Sugiura T. Physiological roles of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Biofactors 2009;35:88-97. https://doi.org/10.1002/biof.18
- Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997;77:299-318. https://doi.org/10.1016/s0306-4522(96)00428-9
- Biegon A, Kerman IA. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. NeuroImage 2001;14:1463-1468. https://doi.org/10.1006/nimg.2001.0939
- Katona I, Rancz EA, Acsady L, et al. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci 2001; 21: 9506-9518. https://doi.org/10.1523/JNEUROSCI.21-23-09506.2001
- Katona I, Urban GM, Wallace M, et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci 2006; 26: 5628-5637. https://doi.org/10.1523/JNEUROSCI.0309-06.2006
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, et al. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 2002; 22:3864-3872. https://doi.org/10.1523/JNEUROSCI.22-10-03864.2002
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 2001;31:453-462. https://doi.org/10.1016/s0896-6273(01)00372-5
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science 2002; 296:678-682. https://doi.org/10.1126/science.1063545
- Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med 2009;11:e3. https://doi.org/10.1017/S1462399409000957
- Mecha M, Feliu A, Carrillo-Salinas FJ, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun 2015;49:233-245. https://doi.org/10.1016/j.bbi.2015.06.002
- Van Sickle MD, Duncan M, Kingsley PJ, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005;310: 329-332. https://doi.org/10.1126/science.1115740
- Brusco A, Tagliaferro PA, Saez T, et al. Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Ann NY Acad Sci 2008;1139:450-457. https://doi.org/10.1196/annals.1432.037
- Onaivi ES, Ishiguro H, Gong JP, et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann NY Acad Sci 2008;1139: 434-449. https://doi.org/10.1196/annals.1432.036
- Maccarrone M, Dainese E, Oddi S. Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci 2010; 35: 601-608. https://doi.org/10.1016/j.tibs.2010.05.008
- Pertwee RG, Howlett AC, Abood ME, et al. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 2010;62:588-631. https://doi.org/10.1124/pr.110.003004
- Aso E, Ferrer I. Cannabinoids for treatment of Alzheimer's disease: moving toward the clinic. Front Pharmacol 2014;5:37. https://doi.org/10.3389/fphar.2014.00037
- Ramírez BG, Blázquez C, Gómez del Pulgar T, et al. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 2005;25: 1904-1913. https://doi.org/10.1523/JNEUROSCI.4540-04.2005
- Solas M, Francis PT, Franco R, et al. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol Aging 2013;34: 805-808. https://doi.org/10.1016/j.neurobiolaging 2012.06.005.
- Benito C, Núñez E, Tolón RM, et al. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci 2003;23:11136-11141. https://doi.org/10.1523/JNEUROSCI.23-35-11136.2003
- Mulder J, Zilberter M, Pasquare SJ, et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011;134: 1041-1060. https://doi.org/10.1093/brain/awr046
- Jung KM, Astarita G, Yasar S, et al. An amyloid β42-dependent deficit in anandamide mobilization is associated with cognitive dysfunction in Alzheimer’s disease. Neurobiol Aging 2012;33: 1522-1532. https://doi.org/10.1016/j.neurobiolaging.2011.03.012
- Hock C, Heese K, Hulette C, et al. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 2000;57:846-851. https://doi.org/10.1001/archneur.57.6.846
- Garzon D, Yu G, Fahnestock M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J Neurochem 2002; 82: 1058-1064. https://doi.org/10.1046/j.1471-4159.2002.01030.x
- Budni J, Bellettini-Santos T, Mina F, et al. The involvement of BDNF, NGF and GDNF in aging and Alzheimer's disease. Aging Dis 2015;6:331-341. https://doi.org/10.14336/AD.2015.0825
- Mohammadi A, Ghasem Amooeian V, Rashidi E. Dysfunction in brain-derived neurotrophic factor signaling pathway and susceptibility to schizophrenia, Parkinson's and Alzheimer's diseases. Curr Gene Ther 2018;18:45-63. https://doi.org/10.2174/1566523218666180302163029
- Numakawa T, Odaka H. Brain-derived neurotrophic factor signaling in the pathophysiology of Alzheimer's disease: beneficial effects of flavonoids for neuroprotection. Int J Mol Sci 2021;22:5719. https://doi.org/10.3390/ijms22115719
- Phillips HS, Hains JM, Armanini M, et al. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991;7:695-702. https://doi.org/10.1016/0896-6273(91)90273-3
- Ferrer I, Marín C, Rey MJ, et al. BDNF and full-length and truncated TrkB expression in Alzheimer disease: implications in therapeutic strategies. J Neuropathol Exp Neurol 1999;58:729-739. https://doi.org/10.1097/00005072-199907000-00007
- Holsinger RM, Schnarr J, Henry P, et al. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res 2000;76:347-354. https://doi.org/10.1016/s0169-328x(00)00023-1
- Wong J, Higgins M, Halliday G, et al. Amyloid beta selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer's disease. Neuroscience 2012;210:363-374. https://doi.org/10.1016/j.neuroscience.2012.02.037
- Fleitas C, Piñol-Ripoll G, Marfull P, et al. proBDNF is modified by advanced glycation end-products in Alzheimer’s disease, and causes neuronal apoptosis by inducing p75 neutrophin receptor processing. Mol Brain 2018;11:68. https://doi.org/10.1186/s13041-018-0411-6
- Ye X, Tai W, Zhang D. The early events of Alzheimer's disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol Aging 2012;33:1122.e1-10. https://doi.org/10.1016/j.neurobiolaging.2011.11.004
- Hellweg R, Gericke CA, Jendroska K, et al. NGF content in the cerebral cortex of non-demented patients with amyloid-plaques and in symptomatic Alzheimer’s disease. Int J Dev Neurosci 1998; 16:787-794. https://doi.org/10.1016/s0736-5748(98)00088-4
- Mufson EJ, Kordower JH. Nerve growth factor receptor expressing human basal forebrain neurons: pathologic alterations in Alzheimer's and Parkinson's disease. Prog Clin Biol Res 1989;317:401-414. PMID: 2557638.
- Fahnestock M, Michalski B, Xu B, et al. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci 2001; 18:210-220. https://doi.org/10.1006/mcne.2001.1016
- Peng S, Wuu J, Mufson EJ, et al. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol 2004; 63:641-649. https://doi.org/10.1093/jnen/63.6.641
- Pedraza CE, Podlesniy P, Vidal N, et al. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol 2005; 166:533-543. https://doi.org/10.1016/S0002-9440(10)62275-4
- Podlesniy P, Kichev A, Pedraza C, et al. Pro-NGF from Alzheimer’s disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am J Pathol 2006; 169:119-131. https://doi.org/10.2353/ajpath.2006.050787
- Al-Shawi R, Hafner A, Olsen J, et al. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur J Neurosci 2008; 27:2103-2114. https://doi.org/10.1111/j.1460-9568.2008.06152.x
- Lin JH, Walter P, Yen TSB. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 2008;3:399-425. https://doi.org/10.1146/annurev.pathmechdis.3.121806.151434
- Kozutsumi Y, Segal M, Normington K, et al. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1998;332:462-464. https://doi.org/10.1038/332462a0
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173-194. https://doi.org/10.1146/annurev-pathol-012513-104649
- Hoozemans JJM, Veerhuis R, Van Haastert ES, et al. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 2005; 110:165-172. https://doi.org/10.1007/s00401-005-1038-0
- Hoozemans JJ, van Haastert ES, Nijholt DA, et al. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 2009;174:1241-1251. https://doi.org/10.2353/ajpath.2009.080814
- Huang HC, Tang D, Lu SY, et al. Endoplasmic reticulum stress as a novel neuronal mediator in Alzheimer's disease. Neurol Res 2015;37:366-374. https://doi.org/10.1179/1743132814Y.0000000448
- Li JQ, Yu JT, Jiang T, Tan L. Endoplasmic reticulum dysfunction in Alzheimer’s disease. Mol Neurobiol 2015;51:383-395. https://doi.org/10.1007/s12035-014-8695-8
- Takuma K, Yan SS, Stern DM, et al. Mitocondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J Pharmacol Sci 2005;97:312-316. https://doi.org/10.1254/jphs cpj04006x.
- Santos LE, Ferreira ST. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer's disease. Neuropharmacology 2018;136:350-360. https://doi.org/10.1016/j.neuropharm.2017.11.016
- Salminen A, Kaarniranta K, Kauppinen A. ER stress activates immunosuppressive network: implications for aging and Alzheimer's disease. J Mol Med 2020;98:633-650. https://doi.org/10.1007/s00109-020-01904-z
- Liu XJ, Wei J, Shang YH, et al. Modulation of AβPP and GSK3β by endoplasmic reticulum stress and involvement in Alzheimer's disease. J Alzheimers Dis 2017;57:1157-1170. https://doi.org/10.3233/JAD-161111
- Uddin MS, Mamun A, Rahman MA, et al. Emerging proof of protein misfolding and interactions in multifactorial Alzheimer's disease. Curr Top Med Chem 2020;20:2380-2390. https://doi.org/10.2174/1568026620666200601161703
- Uddin MS, Tewari D, Sharma G, et al. Molecular mechanisms of ER stress and UPR in the pathogenesis of Alzheimer's disease. Mol Neurobiol 2020;57:2902-2919. https://doi.org/10.1007/s12035-020-01929-y
- Nijholt DA, de Graaf TR, van Haastert ES, et al. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer's disease. Cell Death Differ 2011;18:1071-1081. https://doi.org/10.1038/cdd.2010.176
- Scheper W, Nijholt DA, Hoozemans JJ. The unfolded protein response and proteostasis in Alzheimer disease: preferential activation of autophagy by endoplasmic reticulum stress. Autophagy 2011;7:910-911. https://doi.org/10.4161/auto.7.8.15761
- Cai Y, Arikkath J, Yang L, et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016;12:225-244. https://doi.org/10.1080/15548627.2015.1121360
- Nandi D, Tahiliani P, Kumar A, et al. The ubiquitin-proteasome system. J Biosci 2006;31:137-155. https://doi.org/10.1007/BF02705243
- Wang Y, Le WD. Autophagy and ubiquitin proteasome system. Adv Exp Med Biol 2019;1206:527-550. https://doi.org/10.1007/978-981-15-0602-4_25
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy 2018;14:207-215. https://doi.org/10.1080/15548627.2017.1378838
- Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem 2018; 293:5414-5424. https://doi.org/10.1074/jbc.R117.818237
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93. https://doi.org/10.1146/annurev-genet-102808-114910
- Mehrpour M, Esclatine A, Beau I, et al. Overview of macroautophagy regulation in mammalian cells. Cell Res 2010; 20:748-762. https://doi.org/10.1038/cr.2010.82
- Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 2011; 14:2201-2214. https://doi.org/10.1089/ars.2010.3482
- Cataldo AM, Paskevich PA, Kominami E, et al. Lysosomal hydrolases of different classes are abnormally distributed in brains of patients with Alzheimer disease. Proc Natl Acad Sci USA 1991;88:10998-101002. https://doi.org/10.1073/pnas.88.24.10998
- Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy—a novel b-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 2005;171:87-98. https://doi.org/10.1083/jcb.200505082
- Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immunoelectron microscopy study. J Neuropathol Exp Neurol 2005;64:113-122. https://doi.org/10.1093/jnen/64.2.113
- Barrachina M, Maes T, Buesa C, et al. Up-regulation of lysosome associated-membrane protein 1 (Lamp-1) in Alzheimer’s disease. Neuropathol Appl Neurobiol 2006;32:505-516. https://doi.org/10.1111/j.1365-2990.2006.00756.x
- Cecarini V, Ding Q, Keller JN. Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res 2007; 41: 673-680. https://doi.org/10.1080/10715760701286159
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 2008; 118:2190–2199. https://doi.org/10.1172/JCI33585
- Lipinski MM, Zheng B, Lu T, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci USA 2010;107:14164-14169. https://doi.org/10.1073/pnas.1009485107
- Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 2014;112:24-49. https://doi.org/10.1016/j.pneurobio.2013.10.004
- Uddin MS, Stachowiak A, Mamun AA, et al. Autophagy and Alzheimer's disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci 2018;10:04. https://doi.org/10.3389/fnagi.2018.00004
- Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol 2018;28:3-13. https://doi.org/10.1111/bpa.12545
- Pradeepkiran JA, Reddy PH. Defective mitophagy in Alzheimer's disease. Ageing Res Rev 2020;64:101191. https://doi.org/10.1016/j.arr.2020.101191
- Salminen A, Kaarniranta K, Kauppinen A, et al. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog Neurobiol 2013;106-107:33-54. https://doi.org/10.1016/j.pneurobio.2013.06.002
- Johnston-Carey HK, Pomatto LC, Davies KJ.The Immunoproteasome in oxidative stress, aging, and disease. Crit Rev Biochem Mol Biol 2015;51:268-281. https://doi.org/10.3109/10409238 2016.1172554.
- Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013; 105: 49–59. https://doi.org/10.1016/j.pneurobio 2013.03.001.
- Riederer BM, Leuba G, Vernay A, et al. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med 2011;236:268-276. https://doi.org/10.1258/ebm.2010.010327
- Tramutola A, Di Domenico F, Barone E, et al. Polyubiquitinylation profile in Down syndrome brain before and after the development of Alzheimer neuropathology. Antioxid Redox Signal 2017;26:280-298. https://doi.org/10.1089/ars.2016.6686
- Weng FL, He L. Disrupted ubiquitin proteasome system underlying tau accumulation in Alzheimer's disease. Neurobiol Aging 2021;99:79-85. https://doi.org/10.1016/j.neurobiolaging.2020.11.015
- Ferrer I, Santpere G, van Leeuwen FW. Argyrophilic grain disease. Brain 2008;131:1416-1432. https://doi.org/10.1093/brain/awm305
- Tank EM, True HL. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet 2009;5:e1000382.
- Chadwick L, Gentle L, Strachan J, et al. Review: unchained maladie - a reassessment of the role of Ubb(+1) -capped polyubiquitin chains in Alzheimer's disease. Neuropathol Appl Neurobiol 2012;38:118-131. https://doi.org/10.1111/j.1365-2990.2011.01236.x
- Gentier RJ, van Leeuwen FW. Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer's disease. Front Mol Neurosci 2015;8:47. https://doi.org/10.3389/fnmol.2015.00047
- van Tijn P, Dennissen FJ, Gentier RJ, et al. Mutant ubiquitin decreases β-amyloid plaque formation in a transgenic mouse model of Alzheimer’s disease. Neurochem Int 2012;61:739-748. https://doi.org/10.1016/j.neuint.2012.07.007
- Verheijen BM, Stevens JAA, Gentier RJG, et al. Paradoxical effects of mutant ubiquitin on Aβ plaque formation in an Alzheimer mouse model. Neurobiol Aging 2018;72:62-71. https://doi.org/10.1016/j.neurobiolaging.2018.08.011
- Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol 2004;14:11-20. https://doi.org/10.1111/j.1750-3639.2004.tb00493.x
- Aso E, Lomoio S, López-González I, et al. Amyloid generation and dysfunctional immunoprotasome activation with disease progression in animal model of familial Alzheimer’s disease. Brain Pathol 2012;22:636-653. https://doi.org/10.1111/j.1750-3639.2011.00560.x
- Orre M, Kamphuis W, Dooves S, et al. Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 2013;136: 1415-1431. https://doi.org/10.1093/brain/awt083
- Grimm S, Ott C, Hörlacher M, et al. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem J 2012;448:127-139. https://doi.org/10.1042/BJ20120298
- Thal DR, Del Tredici K, Ludolph AC, et al. Stages of granulovacuolar degeneration: their relation to Alzheimer's disease and chronic stress response. Acta Neuropathol 2011;122:577-589. https://doi.org/10.1007/s00401-011-0871-6
- Yamazaki Y, Matsubara T, Takahashi T, et al. Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PLoS One. 2011;6:e26996. https://doi.org/10.1371/journal.pone.0026996
- Funk KE, Mrak RE, Kuret J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol Appl Neurobiol 2011;37:295-306. https://doi.org/10.1111/j.1365-2990.2010.01135.x
- Köhler C. Granulovacuolar degeneration: a neurodegenerative change that accompanies tau pathology. Acta Neuropathologica 2016; 132: 339-359. https://doi.org/10.1007/s00401-016-1562-0
- Wiersma VI, van Ziel AM, Vazquez‑Sanchez S, et al. Granulovacuolar degeneration bodies are neuron‑selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathologica 2019;138:943-970. https://doi.org/10.1007/s00401-019-02046-4
- Hondius DC, Koopmans F, Leistner C, et al. The proteome of granulovacuolar degeneration and neurofibrillary tangles in Alzheimer’s disease. Acta Neuropathol 2021; 141: 341-358. https://doi.org/10.1007/s00401-020-02261-4
- Hunter S, Minett T, Polvikoski T, et al. Re-examining tau-immunoreactive pathology in the population: granulovacuolar degeneration and neurofibrillary tangles. Alzheimers Res Ther 2015;7:57. https://doi.org/10.1186/s13195-015-0141-2
- Rudy CC, Hunsberger HC, Weitzner DS, et al. The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer's disease. Aging Dis 2015;6:131-148. https://doi.org/10.14336/AD.2014.0423
- Garwood CJ, Radcliffe LE, Simpson JE, et al. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: the supporting player with a central role. Neuropathol Appl Neurobiol 2017; 43:281-298. https://doi.org/10.1111/nan.12338
- Ferrer I. Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol 2017;27:645-674. https://doi.org/10.1111/bpa.12538
- Verkhratsky A, Zorec R, Rodriguez-Arellano JJ, et al. Neuroglia in Ageing. Adv Exp Med Biol 2019;1175:181-197. https://doi.org/10.1007/978-981-13-9913-8_8
- Price BR, Johnson LA, Norris CM. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer's disease. Ageing Res Rev 2021;68:101335. https://doi.org/10.1016/j.arr.2021.101335
- Phillips EC, Croft CL, Kurbatskaya K, et al. Astrocytes and neuroinflammation in Alzheimer's disease. Biochem Soc Trans 2014; 42: 1321-1325. https://doi.org/10.1042/BST20140155
- Heneka MT, Carson MJ, El Khoury J, et al. O, Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14: 388-405. https://doi.org/10.1016/S1474-4422(15)70016-5
- De Strooper BD, Karran E. The cellular phase of Alzheimer’s disease. Cell 2016;164: 603-615. https://doi.org/10.1016/j.cell.2015.12.056
- Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541: 481-487. https://doi.org/10.1038/nature21029
- Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology 2019;27:663-677. https://doi.org/10.1007/s10787-019-00580-x
- Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev 2018;98:239-389. https://doi.org/10.1152/physrev.00042.2016
- Verkhratsky A, Parpura V, Li B, et al. Astrocytes: the housekeepers and guardians of the CNS. Adv Neurobiol 2021;26:21-53. https://doi.org/10.1007/978-3-030-77375-5_2
- Cotrina ML, Nedergaard M. Astrocytes in the aging brain. J Neurosci Res 2002;67:1-10. https://doi.org/10.1002/jnr.10121
- Porchet R, Probst A, Bouras C, et al. Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer's disease. Proteomics 2003;3: 1476-1485. https://doi.org/10.1002/pmic.200300456
- Lynch AM, Murphy KJ, Deighan BF, et al. The impact of glial activation in the aging brain. Aging Dis 2010;1: 262-278. PMID: 22396865.
- Salminen A, Ojala J, Kaarniranta K, et al. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci 2011;34: 3-11. https://doi.org/10.1111/j.1460-9568.2011.07738.x
- Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging 2009;30:337-352. https://doi.org/10.1016/j.neurobiolaging 2007.07.015.
- Popescu BO, Toescu EC, Popescu LM, et al. Blood-brain barrier alterations in ageing and dementia. J Neurol Sci 2009;283: 99-106. https://doi.org/10.1016/j.jns.2009.02.321
- Silverberg GD, Messier AA, Miller MC, et al. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J Neuropathol Exp Neurol 2010;69: 1034-1043. https://doi.org/10.1097/NEN.0b013e3181f46e25
- Silverberg GD, Miller MC, Messier AA, et al. Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol 2010b;69: 98-108. https://doi.org/10.1097/NEN.0b013e3181c8ad2f
- Pérez E, Barrachina M, Rodriguez A, et al. Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer's disease. Brain Res 2007;1128:164-174. https://doi.org/10.1016/j.brainres 2006.09.109.
- Zeppenfeld DM, Simon M, Haswell JD, et al. Association of perivascular localization of Aquaporin-4 with cognition and alzheimer disease in aging brains. JAMA Neurol 2017;74:91-99. https://doi.org/10.1001/jamaneurol.2016.4370
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2005;2: 22. https://doi.org/10.1186/1742-2094-2-22
- Rodríguez JJ, Olabarria M, Chvatal A, et al. Astroglia in dementia and Alzheimer's disease. Cell Death Differ 2009;16: 378-385. https://doi.org/10.1038/cdd.2008.172
- Rodríguez-Arellano JJ, Parpura V, Zorec R, et al. Astrocytes in physiological aging and Alzheimer's disease. Neuroscience 2016;323:170-182. https://doi.org/10.1016/j.neuroscience.2015.01.007
- Verkhratsky A, Rodríguez JJ, Parpura V. Neuroglia in ageing and disease. Cell Tissue Res 2014;357: 493-503. https://doi.org/10.1007/s00441-014-1814-z
- Verkhratsky A, Zorec R, Rodríguez JJ, et al. Astroglia dynamics in ageing and Alzheimer's disease. Curr Opin Pharmacol 2016;26: 74-79. https://doi.org/10.1016/j.coph.2015.09.011
- Nagy JI, Li W, Hertzberg EL, Marotta CA. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer's disease. Brain Res 1996;717: 173-178. https://doi.org/10.1016/0006-8993(95)01526-4
- Kuchibhotla KV, Lattarulo CR, Hyman BT, et al. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009; 323: 1211-1215. https://doi.org/10.1126/science.1169096
- Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 2004;1742: 81-87. https://doi.org/10.1016/j.bbamcr.2004.09.006
- Simpson JE, Ince PG, Shaw PJ, et al. Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer's pathology and APOE genotype. Neurobiol Aging 2011;32: 1795-1807. https://doi.org/10.1016/j.neurobiolaging.2011.04.013
- Galea E, Weinstock LD, Larramona-Arcas R, et al. Multi-transcriptomic analysis points to early organelle dysfunction in human astrocytes in Alzheimer's disease. Neurobiol Dis 2022;166:105655. https://doi.org/10.1016/j.nbd.2022.105655
- Simpson JE, Ince PG, Lace G, et al. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 2010;31: 578-590. https://doi.org/10.1016/j.neurobiolaging 2008.05.015.
- Kamphuis W, Middeldorp J, Kooijman L, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiol Aging 2014;35: 492-510. https://doi.org/10.1016/j.neurobiolaging.2013.09.035
- Beach TG, McGeer EG. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer's disease visual cortex. Brain Res 1988;463: 357-361. https://doi.org/10.1016/0006-8993(88)90410-6
- Yin KJ, Cirrito JR, Yan P, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci 2006;26:10939-10948. https://doi.org/10.1523/JNEUROSCI 2085-06.2006.
- Yang Y, Estrada EY, Thompson JF, et al. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 2007;27:697-709. https://doi.org/10.1038/sj.jcbfm.9600375
- Miners JS, van Helmond Z, Kehoe PG, et al. Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol 2010;20:794-802. https://doi.org/10.1111/j.1750-3639.2010.00375.x
- Carrano A, Hoozemans JJ, van der Vies SM, et al. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis 2012;10:329-331. https://doi.org/10.1159/000334916
- Han BH, Zhou ML, Johnson AW, et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci USA 2015;112:E881–890. https://doi.org/10.1073/pnas.1414930112
- Funato H, Yoshimura M, Yamazaki T, et al. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol 1988;152: 983-992. PMID: 9546359.
- Guénette SY. Astrocytes: a cellular player in Abeta clearance and degradation. Trends Mol Med 2003;9: 279-280. https://doi.org/10.1016/s1471-4914(03)00112-6
- Nagele RG, Wegiel J, Venkataraman V, et al. Contribution of glial cells to the development of amyloid plaques in Alzheimer's disease. Neurobiol Aging 2004;25: 663-674. https://doi.org/10.1016/j.neurobiolaging 2004.01.007.
- Thal DR, Schultz C, Dehghani F, et al. Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol 2000;100: 608-617. https://doi.org/10.1007/s004010000242
- Koistinaho M, Lin S, Wu X, Esterman M, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 2004;10: 719-726. https://doi.org/10.1038/nm1058
- Yan P, Hu X, Song H, Yin K, et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem 2006;281: 24566-24574. https://doi.org/10.1074/jbc.M602440200
- Xiao Q, Yan P, Ma X, et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci 2014;34: 9607-9620. https://doi.org/10.1523/JNEUROSCI.3788-13.2014
- Basak JM, Verghese PB, Yoon H, et al. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J Biol Chem 2012; 287:13959-13971. https://doi.org/10.1074/jbc.M111.288746
- Rossner S, Lange-Dohna C, Zeitschel U, et al. Alzheimer's disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 2005;92: 226-234. https://doi.org/10.1111/j.1471-4159.2004.02857.x
- Zhao J, O'Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation 2011;8: 150. https://doi.org/10.1186/1742-2094-8-150
- Sanchez-Mico MV, Jimenez S, Gomez-Arboledas A, et al. Amyloid beta impairs phagocytosis of dystrophic synapses by astrocytes in Alzheier’s disease. Glia 2021;69:997-1011. https://doi.org/10.1002/glia.23943
- Grolla AA, Sim JA, Lim D, et al. Amyloid-β and Alzheimer's disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis 2013; 4: e623. https://doi.org/10.1038/cddis.2013.145
- Lim D, Ronco V, Grolla AA, et al. Glial calcium signalling in Alzheimer's disease. Rev Physiol Biochem Pharmacol 2014;167: 45-65. https://doi.org/10.1007/112_2014_19
- Verkhratsky A, Rodríguez-Arellano JJ, Parpura V, et al. Astroglial calcium signalling in Alzheimer's disease. Biochem Biophys Res Commun 2017;483:1005-1012. https://doi.org/10.1016/j.bbrc.2016.08.088
- Talantova M, Sanz-Blasco S, Zhang X, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA 2013;110: E2518-2527. https://doi.org/10.1073/pnas.1306832110
- Lim D, Iyer A, Ronco V, et al. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013;61: 1134-1145. https://doi.org/10.1002/glia.22502
- Verkhratsky A, Chvátal A. NMDA receptors in astrocytes. Neurochem Res 2020;45:122-133. https://doi.org/10.1007/s11064-019-02750-3
- Lauderback CM, Hackett JM, Huang FF, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem 2001;78: 413-416. https://doi.org/10.1046/j.1471-4159.2001.00451.x
- Scott HA, Gebhardt FM, Mitrovic AD, et al. Glutamate transporter variants reduce glutamate uptake in Alzheimer's disease. Neurobiol Aging 2011;32: 553.e1-11. https://doi.org/10.1016/j.neurobiolaging 2010.03.008.
- Woltjer RL, Duerson K, Fullmer JM, et al. Aberrant detergent-insoluble excitatory amino acid transporter 2 accumulates in Alzheimer disease. J Neuropathol Exp Neurol 2010;69: 667-676. https://doi.org/10.1097/NEN.0b013e3181e24adb
- Masliah E, Alford M, DeTeresa R, et al. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 1996;40: 759-766. https://doi.org/10.1002/ana.410400512
- Abdul HM, Sama MA, Furman JL, et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci 2009;29:12957-12969. https://doi.org/10.1523/JNEUROSCI.1064-09.2009
- Orre M, Kamphuis W, Osborn LM, Jansen AH, et al. Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 2014;35: 2746-2760. https://doi.org/10.1016/j.neurobiolaging 2014.06.004.
- Dansokho C, Heneka MT. Neuroinflammatory responses in Alzheimer’s disease. J Neural Transm 2018;125:771-779. https://doi.org/10.1007/s00702-017-1831-7
- Tomimoto H, Akiguchi I, Wakita H, et al. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol 1997;94:146-152. https://doi.org/10.1007/s004010050686
- Schipper HM, Bennett DA, Liberman A, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging 2006;27:252-261. https://doi.org/10.1016/j.neurobiolaging.2005.01.016
- Assaraf MI, Diaz Z, Liberman A, et al. Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J Neuropathol Exp Neurol 2007;66:389-398. https://doi.org/10.1097/nen.0b013e3180517b28
- Owen JB, Di Domenico F, Sultana R, et al. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. J Proteome Res 2009;8: 471-482. https://doi.org/10.1021/pr800667a
- Serrano-Pozo A, Gomez-Isla T, Growdon JH, et al. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 2013;182:2332-2344. https://doi.org/10.1016/j.ajpath.2013.02.031
- Perez-Nievas BG, Serrano-Pozo A. Deciphering the astrocyte reaction in alzheimer’s disease. Front Aging Neurosci 2018;10:114. https://doi.org/10.3389/fnagi.2018.00114
- Batiuk MY, Martirosyan A, Wahis J, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun 2020;11: 1220. https://doi.org/10.1038/s41467-019-14198-8
- Sofroniew MV. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol 2020;41:758-770. https://doi.org/10.1016/j.it.2020.07.004
- Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci 2012;32:6391-6410. https://doi.org/10.1523/JNEUROSCI.6221-11.2012
- Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions and future directions. Nat Neurosci 2021;24:312-325. https://doi.org/10.1038/s41593-020-00783-4
- Haxby JV, Grady CL, Koss E, et al. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch Neurol 1990;47: 753-760. https://doi.org/10.1001/archneur.1990.00530070043010
- Small GW, Ercoli LM, Silverman DH, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA 2000;97:6037-6042. https://doi.org/10.1073/pnas.090106797
- de Leon MJ, Convit A, Wolf OT, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc Natl Acad Sci USA 2001;98:10966-10971. https://doi.org/10.1073/pnas.191044198
- Mosconi L, De Santi S, Li J, et al. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging 2008;29:676-692. https://doi.org/10.1016/j.neurobiolaging.2006.12.008
- van Hall G, Stromstad M, Rasmussen P, et al. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab 2009;29:1121-1129. https://doi.org/10.1038/jcbfm.2009.35
- Wyss MT, Jolivet R, Buck A, et al. In vivo evidence for lactate as a neuronal energy source. J Neurosci 2011;31:7477-7485. https://doi.org/10.1523/JNEUROSCI.0415-11.2011
- Proia P, Di Liegro CM, Schiera G, et al. Lactate as a metabolite and a regulator in the central nervous system. Int J Mol Sci 2016;17:1450. https://doi.org/10.3390/ijms17091450
- Alberini CM, Cruz E, Descalzi G, et al. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018;66:1244-1262. https://doi.org/10.1002/glia.23250
- Zhang M, Cheng X, Dang R, et al. Lactate deficit in an Alzheimer disease mouse model: the relationship with neuronal damage. J Neuropathol Exp Neurol 2018;77:1163-1167. https://doi.org/10.1093/jnen/nly102
- Le Foll C, Levin BE. Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol 2016;310:R1186-1192. https://doi.org/10.1152/ajpregu.00113.201 .
- Guzman M, Blazquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab 2001;12:169-173. https://doi.org/10.1016/s1043-2760(00)00370-2
- Panov A, Orynbayeva Z, Vavilin V, et al. Fatty acids in energy metabolism of the central nervous system. Biomed Res Int 2014;2014:472459. https://doi.org/10.1155/2014/472459
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011;14:724-38. https://doi.org/10.1016/j.cmet.2011.08.016
- Steele ML, Robinson SR. Reactive astrocytes give neurons less support: implications for Alzheimer's disease. Neurobiol Aging 2012;33:423.e1-13. https://doi.org/10.1016/j.neurobiolaging.2010.09.018
- Zulfiqar S, Garg P, Nieweg K. Contribution of astrocytes to metabolic dysfunction in the Alzheimer's disease brain. Biol Chem 2019;400:1113-1127. https://doi.org/10.1515/hsz-2019-0140
- Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 2016;173:649-665. https://doi.org/10.1111/bph.13139
- Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 2016;53:1181-1194. https://doi.org/10.1007/s12035-014-9070-5
- Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 2016;19:987-991. https://doi.org/10.1038/nn.4338
- Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol 2010; 119:89-105. https://doi.org/10.1007/s00401-009-0622-0
- Svahn AJ, Becker TS, Graeber MB. Emergent properties of microglia. Brain Pathol 2014;24:665-670. https://doi.org/10.1111/bpa.12195
- Hashemiaghdam A, Mroczek M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer's disease. J Neuroimmunol 2020;341:577185. https://doi.org/10.1016/j.jneuroim.2020.577185
- Streit WJ, Samkons NW, Kuhns AJ, et al. Dystrophic microglia in the aging human brain. Glia 2004;45:208-212. https://doi.org/10.1002/glia.10319
- Yuan C, Aierken A, Xie Z, et al. The age-related microglial transformation in Alzheimer's disease pathogenesis. Neurobiol Aging 2020;92:82-91. https://doi.org/10.1016/j.neurobiolaging.2020.03.024
- Saez-Atienzar S, Masliah E. Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat Rev Neurosci 2020;21:433-444. https://doi.org/10.1038/s41583-020-0325-z
- Albrecht DS, Sagare A, Pachicano M, et al. Early neuroinflammation is associated with lower amyloid and tau levels in cognitively normal older adults. Brain Behav Immun 2021;94:299-307. https://doi.org/10.1016/j.bbi.2021.01.010
- Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer's disease. Brain 2018;141:2740-2754. https://doi.org/10.1093/brain/awy188
- Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging 2000;21:383-421. https://doi.org/10.1016/s0197-4580(00)00124-x
- Streit WJ, Conde JR, Harrison JK. Chemokines and Alzheimer’s disease. Neurobiol Aging 2001;22:909-913. https://doi.org/10.1016/s0197-4580(01)00290-1
- McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimers Dis 2010;19:355-361. https://doi.org/10.3233/JAD-2010-1219
- Morimoto K, Horio J, Satoh H, et al. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients, compared to the brains of non-demented patients with and without increasing AD pathology. J Alzheimers Dis 2011;25:59-76. https://doi.org/10.3233/JAD-2011-101815
- Nagae T, Araki K, Shimoda Y, et al. Cytokines and cytokine receptors involved in the pathogenesis of Alzheimer's disease. J Clin Cell Immunol 2016;7:441. https://doi.org/10.4172/2155-9899.1000441
- Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathol 2013;126:461-477. https://doi.org/10.1007/s00401-013-1182-x
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014;14:463-477. https://doi.org/10.1038/nri3705
- Jorda A, Campos-Campos J, Iradi A, et al. The role of chemokines in Alzheimer's disease. Endocr Metab Immune Disord Drug Targets 2020;20:1383-1390. https://doi.org/10.2174/1871530320666200131110744
- López-González I, Schlüter A, Aso E, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol 2015;74: 319-344. https://doi.org/10.1097/NEN.0000000000000176
- López-González I, Aso E, Carmona M, et al. Neuroinflammatory gene regulation, mitochondrial function, oxidative stress, and brain lipid modifications with disease progression in Tau P301S transgenic mice as a model of frontotemporal lobar degeneration-Tau. J Neuropathol Exp Neurol 2015;74: 975-999. https://doi.org/10.1097/NEN.0000000000000241
- Loving BA, Bruce KD. Lipid and lipoprotein metabolism in microglia. Front Physiol. 2020;11:393. https://doi.org/10.3389/fphys.2020.00393
- McGeer PL, McGeer EG. The amyloid cascade/inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol 2013;126:479-497. https://doi.org/10.1007/s00401-013-1177-7
- Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci 2014;124:307-321. https://doi.org/10.3109/00207454.2013.833510
- Bamberger ME, Harris ME, McDonald DR, et al. A cell surface receptor complex for fibrillary-amyloid mediates microglial activation. J Neurosci 2003;23:2665-2674. https://doi.org/10.1523/JNEUROSCI.23-07-02665.2003
- Mandrekar S, Jiang Q, Lee CY, et al. Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci 2009;29:4252-4262. https://doi.org/10.1523/JNEUROSCI.5572-08.2009
- Carroll MC. The complement system in B cell regulation. Mol Immunol 2004;41:141-146. https://doi.org/10.1016/j.molimm.2004.03.017
- Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol 2004;41:1089-1098. https://doi.org/10.1016/j.molimm.2004.06.011
- Reichwald J, Danner S, Wiederhold KH, et al. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation 2009; 6:35. https://doi.org/10.1186/1742-2094-6-35
- Pan XD, Zhu YG, Lin N, et al. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: implications for Alzheimer’s disease. Mol Neurodegener 2011;6:45. https://doi.org/10.1186/1750-1326-6-45
- Vogels T, Murgoci AN, Hromádka T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun 2019;7:109. https://doi.org/10.1186/s40478-019-0754-y
- Sheffield LG, Marquis JG, Berman NE. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett 2000;285: 165-168. https://doi.org/10.1016/s0304-3940(00)01037-5
- Serrano-Pozo A, Mielke ML, Gómez-Isla T, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol 2011;179:1373-1384. https://doi.org/10.1016/j.ajpath.2011.05.04
- Perea JR, Bolós M, Avila J. Microglia in Alzheimer’s disease in the context of tau pathology. Biomolecules 2020;10:1439. https://doi.org/10.3390/biom10101439
- Zhou R, Ji B, Kong Y, et al. PET imaging of neuroinflammation in Alzheimer's disease. Front Immunol 2021;12:739130. https://doi.org/10.3389/fimmu.2021.739130
- Pascoal TA, Benedet AL, Ashton NJ, et al. Microglial activation and tau propagate jointlyacross Braak stages. Nat Med 2021;27:1592-1599. https://doi.org/10.1038/s41591-021-01456-w
- Vogels T, Leuzy A, Cicognola C, et al. Propagation of tau pathology: integrating insights from postmortem and in vivo studies. Biol Psychiatry 2020; 87: 808-818. https://doi.org/10.1016/j.biopsych.2019.09.019
- Gerrits E, Brouwer N, Kooistra SM, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer's disease. Acta Neuropathol 2021;141:681-696. https://doi.org/10.1007/s00401-021-02263-w
- Wilcock DM, Hurban J, Helman AM, et al. Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol Aging 2015;36:2468-2474. https://doi.org/10.1016/j.neurobiolaging.2015.05.016
- Flores-Aguilar L, Iulita MF, Kovecses O, et al. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 2020;143:3653-3671. https://doi.org/10.1093/brain/awaa326
- Ahmed MM, Johnson NR, Boyd TD, et al. Innate immune system activation and neuroinflammation in Down syndrome and neurodegeneration: therapeutic targets or partners? Front Aging Neurosci 2021;13:718426. https://doi.org/10.3389/fnagi.2021.718426
- Barroeta-Espar I, Weinstock LD, Perez-Nievas BG, et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s disease. Neurobiol Dis 2019;121:327-337. https://doi.org/10.1016/j.nbd.2018.10.009
- Yang J, Wise L, Fukuchi KI. TLR4 Cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer's disease. Front Immunol 2020;11:724. https://doi.org/10.3389/fimmu.2020.00724
- Hanslik KL, Ulland TK. The role of microglia and the Nlrp3 inflammasome in Alzheimer's disease. Front Neurol 2020;11:570711. https://doi.org/10.3389/fneur.2020.570711
- Zhang Y, Zhao Y, Zhang J, et al. Mechanisms of NLRP3 inflammasome activation: its role in the treatment of Alzheimer's disease. Neurochem Res 2020;45:2560-2572. https://doi.org/10.1007/s11064-020-03121-z
- Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008;9:857-865. https://doi.org/10.1038/ni.1636
- Lučiūnaitė A, McManus RM, Jankunec M, et al. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem 2020;155:650-661. https://doi.org/10.1111/jnc.14945
- Van Zeller M, Dias D, Sebastião AM, et al. NLRP3 inflammasome: A starring role in amyloid-β- and tau-driven pathological events in Alzheimer's disease. J Alzheimers Dis 2021;83:939-961. https://doi.org/10.3233/JAD-210268
- Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674-678. https://doi.org/10.1038/nature11729
- Stancu IC, Cremers N, Vanrusselt H, et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 2019;137:599-617. https://doi.org/10.1007/s00401-018-01957-y
- Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019;575:669-673. https://doi.org/10.1038/s41586-019-1769-z
- Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117-127. https://doi.org/10.1056/NEJMoa1211851
- Takatori S, Wang W, Iguchi A, Tomita T. Genetic risk factors for Alzheimer disease: emerging roles of microglia in disease pathomechanisms. Adv Exp Med Biol 2019;1118:83-116. https://doi.org/10.1007/978-3-030-05542-4_5
- Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 2021;17:157-172. https://doi.org/10.1038/s41582-020-00435-y
- Egensperger R, Kösel S, von Eitzen U, et al. Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol 1998;8:439-447. https://doi.org/10.1111/j.1750-3639.1998.tb00166.x
- Kloske CM, Dugan AJ, Weekman EM, et al. Inflammatory pathways are impaired in Alzheimer disease and differentially associated with apolipoprotein E status. J Neuropathol Exp Neuro 2021; 80: 922-932. https://doi.org/10.1093/jnen/nlab085
- Singh AK, Mishra G, Maurya A, et al. Role of TREM2 in Alzheimer's disease and its consequences on β-amyloid, tau and neurofibrillary tangles. Curr Alzheimer Res 2019;16:1216-1229. https://doi.org/10.2174/1567205016666190903102822
- Griciuc A, Tanzi RE. The role of innate immune genes in Alzheimer’s disease. Curr Opin Neurol 2021;34: 228-236. https://doi.org/10.1097/WCO.0000000000000911
- Prokop S, Miller KR, Labra SR, et al. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer's disease patient brain samples. Acta Neuropathol 2019;138:613-630. https://doi.org/10.1007/s00401-019-02048-2
- Toomey CE, Heywood W, Benson BC, et al. Investigation of pathology, expression and proteomic profiles in human TREM2 variant postmortem brains with and without Alzheimer's disease. Brain Pathol 2020;30:794-810. https://doi.org/10.1111/bpa.12842
- Ferrer I. Oligodendrogliopathy in neurodegenerative diseases with abnormal protein aggregates: the forgotten pattern. Prog Neurobiol 2018;169:24-54. https://doi.org/10.1016/j.pneurobio.2018.07.004
- Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol 1987;44:21-23. https://doi.org/10.1001/archneur.1987.00520130013009
- Scheltens P, Barkhof F, Leys D, et al. Histopathologic correlates of white matter changes on MRI in Alzheimer's disease and normal aging. Neurology 1995;45:883-888. https://doi.org/10.1212/wnl.45.5.883
- Salat DH, Tuch DS, Greve DN, et al. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol Aging 2005; 26:1215-1227. https://doi.org/10.1016/j.neurobiolaging.12004.09.017
- Holland CM, Smith EE, Csapo I, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 2008; 39: 1127-1133. https://doi.org/10.1161/STROKEAHA.107.497438
- Liu H, Yang Y, Xi Y, et al. Aging of cerebral white matter. Ageing Res Rev 2017;34:64-76. https://doi.org/10.1016/j.arr.2016.11.006
- Marner L, Nyengaard JR, Tang Y, et al. Marked loss of myelinated nerve fibers in the human brain with age. https://doi.org/10.1016/j.arr.2016.11.006
- Erten-Lyons D, Woltjer R, Kaye J, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 2013; 81: 977-983. https://doi.org/10.1016/j.arr.2016.11.006
- Kohama SG, Rosene DL, Sherman LS. Age-related changes in human and non-human primate white matter: from myelination disturbances to cognitive decline. Age 2012; 34: 1093-1110. https://doi.org/10.1007/s11357-011-9357-7
- Bartzokis G, Cummings JL, Sultzer D, et al. White matter structural integrity in healthy aging adults and patients with Alzheimer disease: a magnetic resonance imaging study. Arch Neurol 2003; 60: 393-398. https://doi.org/10.1001/archneur.60.3.393
- Wang L, Goldstein FC, Levey AI, et al. White matter hyperintensities and changes in white matter integrity in patients with Alzheimer’s disease. Neuroradiology 2011;53:373-381. https://doi.org/10.1007/s00234-010-0806-2
- Radanovic M, Pereira FR, Stella F, et al. White matter abnormalities associated with Alzheimer's disease and mild cognitive impairment: a critical review of MRI studies. Expert Rev Neurother 2013; 13:483-493. https://doi.org/10.1586/ern.13.45
- Selnes P, Fjell AM, Gjerstad L, et al. White matter imaging changes in subjective and mild cognitive impairment. Alzheimers Dement 2012;8:S112-S121. https://doi.org/10.1016/j.jalz.2011.07.001
- Molinuevo JL, Ripolles P, Simo M, et al. White matter changes in preclinical Alzheimer's disease: a magnetic resonance imaging diffusion tensor imaging study on cognitively normal older people with positive amyloid beta protein 42 levels. Neurobiol Aging 2014; 35: 2671-2680. https://doi.org/10.1016/j.neurobiolaging.2014.05.027
- Hoy AR, Ly M, Carlsson CM, Okonkwo OC, et al. Microstructural white matter alterations in preclinical Alzheimer's disease detected using free water elimination diffusion tensor imaging. PLoS One 2017; 12: e0173982. https://doi.org/10.1371/journal.pone.0173982
- Bouhrara M, Reiter DA, Bergeron CM, et al. Evidence of demyelination in mild cognitive impairment and dementia using a direct and specific magnetic resonance imaging measure of myelin content. Alzheimers Dement 2018; 14: 998-1004. https://doi.org/10.1016/j.jalz.2018.03.007
- Zhang X, Sun Y, Li W, et al. Characterization of white matter changes along fibers by automated fiber quantification in the early stages of Alzheimer's disease NeuroImage: Clinical 2019; 22: 101723. https://doi.org/10.1016/j.nicl.2019.101723
- Gouw AA, Seewann A, Vrenken H, et al. Heterogeneity of white matter hyperintensities in Alzheimer's disease: post-mortem quantitative MRI and neuropathology. Brain 2008; 131: 3286-3298. https://doi.org/10.1093/brain/awn265
- Ihara M, Polvikoski TM, Hall R, et al. Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 2010; 119: 579-589. https://doi.org/10.1007/s00401-009-0 35-8.
- Rose SE, Chen F, Chalk JB, et al. Loss of connectivity in Alzheimer's disease: an evaluation of white matter tract integrity with colour coded MR diffusion tensor imaging. J Neurol Neurosurg Psychiatry 2000;69:528-530. https://doi.org/10.1136/jnnp.69.4.528
- Bennett IJ, Madden DJ. Disconnected aging: cerebral white matter integrity and age-related differences in cognition. Neuroscience 2014; 276: 187-205. https://doi.org/10.1016/j.neuroscience.2013.11.026
- Desai MK, Sudol KL, Janelsins MC, et al. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 2009; 57: 54-65. https://doi.org/10.1002/glia.20734
- Kamphuis W, Orre M, Kooijman L, et al. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 2012; 60: 615-629. https://doi.org/10.1002/glia.22295
- Dong XX, Zhang HY, Li HY, et al. Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction. Neural Regen Res 2018;13:908-914. https://doi.org/10.4103/1673-5374.232486
- Gu L, Wu D, Tang X, et al. Myelin changes at early stage of 5xFAD mice. Brain Res Bull 2018; 137:285-293. https://doi.org/10.1016/j.brainresbull.2017.12.013
- Scheibel AB, Duong TH, Jacobs R. Alzheimer's disease as a capillary dementia. Ann Med 1989;21:103-107. https://doi.org/10.3109/07853898909149194
- de la Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 2004;3:184-190. https://doi.org/10.1016/S1474-4422(04)00683-0
- de la Torre JC. Alzheimer’s disease is a vasocognopathy: a new term to describe its nature. Neurol Res 2004;26:517-524. https://doi.org/10.1179/016164104225016254
- Henry-Feugeas MC. Alzheimer's disease in late-life dementia: a minor toxic consequence of devastating cerebrovascular dysfunction. Med Hypotheses 2008;70:866-875. https://doi.org/10.1016/j.mehy.2007.07.027
- Østergaard L, Aamand R, Gutiérrez-Jiménez E, et al. The capillary dysfunction hypothesis of Alzheimer's disease. Neurobiol Aging 2013; 34:1018-1031. https://doi.org/10.1016/j.neurobiolaging.2012.09.011
- Scheibel AB, Duong TH, Tomiyasu U. Denervation microangiopathy in senile dementia, Alzheimer type. Alzheimer Dis Assoc Disord 1987;1:19-37. https://doi.org/10.1097/00002093-198701000-00004
- Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 2001;64:575-611. https://doi.org/10.1016/s0301-0082(00)00068-x
- Jellinger K. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002;109; 813-836. https://doi.org/10.1007/s007020200068
- Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983;14:924-928. https://doi.org/10.1161/01.str.14.6.924
- Kalaria RN, Premkumar DR, Pax AB, et al. Production and increased detection of amyloid beta protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroids plexus in Alzheimer's disease. Mol Brain Res 1996;35:58–68. https://doi.org/10.1016/0169-328x(95)00180-z
- Buée L, Hof PR, Bouras C, et al. Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol 1994;87:469-480. https://doi.org/10.1007/BF00294173
- Castellani RJ, Smith MA, Perry G, et al. Cerebral amyloid angiopathy: major contributor or decorative response to Alzheimer's disease pathogenesis. Neurobiol Aging 2004;25:599-602. https://doi.org/10.1016/j.neurobiolaging.2003.12.019
- Thal DR, Griffin WST, de Vos RAI, et al. Cerebral amyloid angioapthy and its relationship to Alzheimer’s disease. Acta Neuropathol 2008;115:599-609. https://doi.org/10.1007/s00401-008-0366-2
- Weller RO, Subash M, Preston SD, et al. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 2008;18:253-266. https://doi.org/10.1111/j.1750-3639.2008.00133.x
- Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s diseaseand their potential impact on therapy. Acta Neuropathol 2009;118:87-102. https://doi.org/10.1007/s00401-009-0498-z
- Soontornniyomkij V, Choi C, Pomakian J, et al. High-definition characterization of cerebral beta-amyloid angiopathy in Alzheimer's disease. Hum Pathol 2010;41:1601-1608. https://doi.org/10.1016/j.humpath.2010.04.011
- Hunter JM, Kwan J, Malek-Ahmadi M, et al. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer's disease. PLoS One 2012;7:e36893. https://doi.org/10.1371/journal.pone.0036893
- Magaki S, Tang Z, Tung S, et al. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol Aging 2018;70:70-77. https://doi.org/10.1016/j.neurobiolaging.2018.06.004
- Jäkel L, De Kort AM, Klijn CJM, et al. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimers Dement 2022;18:10-28. https://doi.org/10.1002/alz.12366
- Apátiga-Pérez R, Soto-Rojas LO, Campa-Córdoba BB, et al. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer's disease. Metab Brain Dis 2022;37:39-50. https://doi.org/10.1007/s11011-021-00814-4
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosc 2004;5:347-360. https://doi.org/10.1038/nrn1387
- Johnson NA, Jahng GH, Weiner MW, Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 2005;234:851-859. https://doi.org/10.1148/radiol.23 3040197.
- Sagare AP, Bell RD, Zhao Z, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 2013;4:2932. https://doi.org/10.1038/ncomms3932
- Franzblau M, Gonzales-Portillo C, Gonzales-Portillo GS, et al. Vascular damage: a persisting pathology common to Alzheimer’s disease and traumatic brain injury. Med Hypotheses 2013;81:842-845. https://doi.org/10.1016/j.mehy.2013.09.012
- Nelson AR, Sweeney MD, Sagare AP, et al. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 2016;1862:887-900. https://doi.org/10.1016/j.bbadis.2015.12.016
- Kisler K, Nelson AR, Montagne A, et al. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 2017;18:419-434. https://doi.org/10.1038/nrn.2017.48
- Leijenaar JF, van Maurik IS, Kuijer JPA, et al. Lower cerebral blood flow in subjects with Alzheimer’s dementia, mild cognitive impairment, and subjective cognitive decline using two-dimensional phase-contrast magnetic resonance imaging. Alzheimers Dement 2017;9:76-83. https://doi.org/10.1016/j.dadm.2017.10.001
- Yamazaki Y, Kanekiyo T. Blood-Brain Barrier dysfunction and the pathogenesis of Alzheimer's disease. Int J Mol Sci 2017;18:1965. https://doi.org/10.3390/ijms18091965
- Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol 2018;17:1016-1024. https://doi.org/10.1016/S1474-4422(18)30318-1
- Ahmad A, Patel V, Xiao J, et al. The role of neurovascular system in neurodegenerative diseases. Mol Neurobiol 2020;57: 4373-4393. https://doi.org/10.1007/s12035-020-02023-z
- Sweeney MD, Montagne A, Sagare AP, et al. Vascular dysfunction: The disregarded partner of Alzheimer's disease. Alzheimers Dement 2019;15:158-167. https://doi.org/10.1016/j.jalz.2018.07.222
- Kurz C, Walker L,Rauchmann BS, et al. Dysfunction of the blood-brain barrier in Alzheimer’s disease: evidence from human studies. Neuropathol Appl Neurobiol Neuropathol Appl Neurobiol 2022;e12782. https://doi.org/10.1111/nan.12782
- Kalaria RN, Pax AB. Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res 1995;705:349-352. https://doi.org/10.1016/0006-8993(95)01250-8
- Verbeek MM, Otte-Holler I, van den Born J, et al. Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer's disease brain. Am J Pathol 1999;155:2115-2125. https://doi.org/10.1016/S0002-9440(10)65529-0
- Berzin TM, Zipser BD, Rafii MS, et al. Agrin and microvascular damage in Alzheimer’s disease. Neurobiol Aging 2000;21:349-355. https://doi.org/10.1016/s0197-4580(00)00121-4
- Grammas P, Yamada M, Zlokovic B. The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 2002;4:217-223. https://doi.org/10.3233/jad-2002-4311
- Ervin JF, Pannell C, Szymansky M, et al. Vascular smooth muscle actin is reduced in Alzheimer disease brain: a quantitative analysis. J Neuropathol Exp Neurol 2004;63:735-741. https://doi.org/10.1093/jnen/63.7.735
- Bailey TL, Rivara CB, Rocher AB, et al. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 2004;26:573-578. https://doi.org/10.1179/016164104225016272
- Kalaria RN, Hedera P. Differential degeneration of the cerebral microvasculature in Alzheimer’s disease. Neuroreport 1995;6:477-480. https://doi.org/10.1097/00001756-199502000-00018
- Wu Z, Guo H, Chow N, Sallstrom J, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005;11:959-965. https://doi.org/10.1038/nm1287
- Hashimura T, Kimura T, Miyakawa T. Morphological changes of blood vessels in the brain with Alzheimer’s disease. Jpn J Psychiatry Neurol 1991;45:661-665. https://doi.org/10.1111/j.1440-1819.1991.tb01187.x
- Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer's disease. J Alzheimers Dis 2013;33:S87-100. https://doi.org/10.3233/JAD-2012-129037
- Ma Q, Zhao Z, Sagare AP, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener 2018;13:57. https://doi.org/10.1186/s13024-018-0286-0
- Lendahl U, Nilsson P, Betsholtz C. Emerging links between cerebrovascular and neurodegenerative diseases-a special role for pericytes. EMBO Rep 2019;20:e48070. https://doi.org/10.15252/embr.201948070
- Nortley R, Korte N, Izquierdo P, et al. β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 2019;365:eaav9518. https://doi.org/10.1126/science.aav9518
- Winkler EA, Sengillo JD, Bell RD, et al. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J Cereb Blood Flow Metab 2012;32:1841-1852. https://doi.org/10.1038/jcbfm.2012.113
- Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 2014;24:371-386. https://doi.org/10.1111/bpa.12152
- Kalback W, Esh C, Castano EM, et al. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer's disease. Neurol Res 2004;26:525-539. https://doi.org/10.1179/016164104225017668
- Shabir O, Berwick J, Francis SE. Neurovascular dysfunction in vascular dementia, Alzheimer's and atherosclerosis. BMC Neurosci 2018;19:62. https://doi.org/10.1186/s12868-018-0465-5
- Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012;485:512-516. https://doi.org/10.1038/nature11087
- Mosconi L, Sorbi S, de Leon MJ, et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med 2006;47:1778–1786. PMID: 17079810.
- Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019; 25: 270-276. https://doi.org/10.1038/s41591-018-0297-y
- Winkler EA, Nishida Y, Sagare AP, et al. GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 2015;18:521-530. https://doi.org/10.1038/nn.3966
- Koepsell H. Glucose transporters in brain in health and disease. Pflugers Arch 2020;472:1299-1343. https://doi.org/10.1007/s00424-020-02441-x
- An Y, Varma VR, Varma S, et al. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement 2018;14:318-329. https://doi.org/10.1016/j.jalz.2017.09.011
- Szu JI, Obenaus A.. Cerebrovascular phenotypes in mouse models of Alzheimer's disease. Cereb Blood Flow Metab 2021;41:1821-1841. https://doi.org/10.1177/0271678X21992462
- Canepa E, Fossati S. Impact of Tau on neurovascular pathology in Alzheimer's disease. Front Neurol 2021;11:573324. https://doi.org/10.3389/fneur.2020.573324
- Blair LJ, Frauen HD, Zhang B, et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun 2015;3:8. https://doi.org/10.1186/s40478-015-0186-2
- Love S, Miners J. Cerebrovascular disease in ageing and Alzheimer's disease Acta Neuropathol 2016;131:645-658. https://doi.org/10.1007/s00401-015-1522-0
- Yu X, Ji C, Shao A. Neurovascular unit dysfunction and neurodegenerative disorders. Front Neurosci 2020;14:334. https://doi.org/10.3389/fnins.2020.00334
- Gordon GR, Choi HB, Rungta RL, et al. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 2008; 456: 745-749. https://doi.org/10.1038/nature07525
- Ipata PL, Camici M, Micheli V, et al. Metabolic network of nucleosides in the brain. Curr Top Med Chem 2011;11:909-922. https://doi.org/10.2174/156802611795347555
- Ansoleaga B, Jove M, Schluter A, et al. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol Aging 2015;36:68-80. https://doi.org/10.1016/j.neurobiolaging.2014.08.004
- Rahman A. The role of adenosine in Alzheimer’s disease. Curr Neuropharmacol 2009; 7: 207-216. https://doi.org/10.2174/157015909789152119
- Carman AJ, Mills JH, Krenz A, et al. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J Neurosci 2011; 31: 13272-13280. https://doi.org/10.1523/JNEUROSCI.3337-11.201
- Alonso-Andrés P, Albasanz JL, Ferrer I, et al. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol 2018;28: 933-946. https://doi.org/10.1111/bpa.12592
- Panza F, Frisardi V, Capurso C, et al. Polyunsaturated fatty acid and S-adenosylmethionine supplementation in predementia syndromes and Alzheimer’s disease: a review. ScientificWorldJournal 2009;9:373-389. https://doi.org/10.1100/tsw.2009.48
- Ruan Q, Ruan J, Zhang W, Qian F, et al. Targeting NAD(+) degradation: The therapeutic potential of flavonoids for Alzheimer's disease and cognitive frailty. Pharmacol Res 2018;128:345-358. https://doi.org/10.1016/j.phrs.2017.08.010
- Erb L, Woods LT, Khalafalla MG, et al. Purinergic signalling in Alzheimer’s disease. Brain Res Bull 2019;151:25-37. https://doi.org/10.1016/j.brainresbull.2018.10.014
- Sharma VK, Singh TG, Singh S. Cyclic nucleotides signaling and phosphodiesterase inhibition: defying Alzheimer's disease. Curr Drug Targets 2020;21:1371-1384. https://doi.org/10.2174/1389450121666200727104728
- Merighi S, Poloni TE, Terrazzan A, et al. Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells 2021;10:1267. https://doi.org/10.3390/cells10051267
- Pan L, Ma Y, Li Y, Wu H, et al. The therapeutic potential of purinergic receptors in Alzheimer's disease and promising therapeutic modulators. Mini Rev Med Chem 2021;21:1288-1302. https://doi.org/10.2174/1389557520999201209211610
- Kundu D, Dubey VK. Purines and pyrimidines: metabolism, function and potential as therapeutic options in neurodegenerative diseases. Curr Protein Pept Sci 2021;22:170-189. https://doi.org/10.2174/1389203721999201208200605
- Allis CD, Caparros ML, Jenuwein T, et al. Epigenetics Second Edit. Cold Springer Arbor Laboratory Press 2015.
- Paro R, Grossniklaus U, Santoro R, Wutz A. Introduction to epigenetics, Springer, 2021
- Wu H, Tao J, Sun YE. Regulation and function of mammalian DNA methylation patterns: a genomic perspective. Brief Funct Genomics. 2012;11:240-250. https://doi.org/10.1093/bfgp/els01
- Qazi TJ, Quan Z, Mir A, Qing H. Epigenetics in Alzheimer's Disease: Perspective of DNA Methylation. Mol Neurobiol 2018;55:1026-1044. https://doi.org/10.1007/s12035-016-0357-6
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997;389: 349-352. https://doi.org/10.1038/38664
- Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 1998; 20: 615-626. https://doi.org/10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H
- de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACS): characterization of the classical HDAC family. Biochem J 2003;370:737-749. https://doi.org/10.1042/BJ20021321
- Torok MS, Grant PA. Histone acetyltransferase proteins contribute to transcriptional processes at multiple levels. Adv Protein Chem 2004;67:181-199. https://doi.org/10.1016/S0065-3233(04)67007-0
- Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol Endocrin 2005;19:563-573. https://doi.org/10.1210/me.2004-0496
- Imhof A. Epigenetic regulators and histone modification. Brief Funct Genomic Proteomic 2006;5:222-227. https://doi.org/10.1093/bfgp/ell030
- Gupta S, Kim SY, Artis S, et al. Histone methylation regulates memory formation. J Neurosci 2010; 30:3589–3599. https://doi.org/10.1523/JNEUROSCI.3732-09.2010
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Rev Genet 2012; 13:343-357. https://doi.org/10.1038/nrg3173
- Morrison LD, Smith DD, Kish SJ. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J Neurochem 1996;67:1328-1331. https://doi.org/10.1046/j.1471-4159.1996.67031328.x
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–935. https://doi.org/10.1126/science.1170116
- Mastroeni D, McKee A, Grover A, et al. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One 2009; 4 e6617. https://doi.org/10.1371/journal.pone.0006617
- Tan L, Shi YG. Tet family proteins and 5- hydroxymethylcytosine in development and disease. Development 2012;139:1895–1902. https://doi.org/10.1242/dev.070771
- Irier HA, Jin P. Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol 2012;31:S42–S48. https://doi.org/10.1089/dna.2011.1565
- Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 2017;18:517-534. https://doi.org/10.1038/nrg.2017.33
- Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2007; 2:e895. https://doi.org/10.1371/journal.pone.0000895
- Wang SC, Oeize B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One 2008; 2008;3:e2698. https://doi.org/10.1371/journal.pone.0002698
- Mastroeni D, Grover A, Delvaux E, et al. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging 2011;32:1161-1180. https://doi.org/10.1016/j.neurobiolaging.2010.08.017
- Bakulski KM, Dolinoy DC, Sartor MA et al. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis 2012;29:571-588. https://doi.org/10.3233/JAD-2012-111223
- Watson CT, Roussos P, Garg P, et al. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med 2016;8:5. https://doi.org/10.1186/s13073-015-0258-8
- Smith AR, Smith RG, Pishva E, et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer's disease. Clin Epigenetics 2019;11:52. https://doi.org/10.1186/s13148-019-0636-y
- Sanchez-Mut JV, Aso E, Panayotis N, et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013;136:3018–3027. https://doi.org/10.1093/brain/awt237
- Sanchez-Mut JV, Heyn H, Vidal E, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry 2016;6:e718. https://doi.org/10.1038/tp.2015.214
- Rao JS, Keleshian VL, Klein S, et al. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry 2012;2: e132. https://doi.org/10.1038/tp.2012.55
- Scarpa S, Fuso A, D’Anselmi F, et al. Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Lett 2003;541:145-148. https://doi.org/10.1016/s0014-5793(03)00277-1
- Fuso A, Seminara L, Cavallaro RA, et al. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci 2005;28:195–204. https://doi.org/10.1016/j.mcn.2004.09.007
- Monti N, Cavallaro RA, Stoccoro A, et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 2020;15: 781-799. https://doi.org/10.1080/15592294.2020.1722917
- Sontag E, Nunbhakdi-Craig V, Sontag J-M, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 2007; 27:2751-2759. https://doi.org/10.1523/JNEUROSCI.3316-06.2007
- Zhou XW, Gustafsson JA, Tanila H, et al. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol Dis 2008;31:386-394. https://doi.org/10.1016/j.nbd.2008.05.013
- Bernstein AI, Lin Y, Street RC, et al. 5-Hydroxymethylation associated epigenetic modifiers of Alzheimer’s disease modulates tau induced neurotoxicity. Hum Mol Genet 2016;25:2437-2450. https://doi.org/10.1093/hmg/ddw109
- Pietrzak M, Rempala G, Nelson PT, et al. Epigenetic silencing of nucleolar rRNA genes in Alzheimer's disease. PLoS One 2011;6:e22585. https://doi.org/10.1371/journal.pone.0022585
- Nagata T, Kobayashi N, Ishii J, et al. Association between DNA methylation of the BDNF promoter region and clinical presentation in Alzheimer's disease. Dement Geriatr Cogn Dis Extra 2015;5:64-73. https://doi.org/10.1159/000375367
- Wilson AG. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol 2008;79:1514-1519. https://doi.org/10.1902/jop.2008.080172
- Smith AR, Smith RG, Condliffe D, et al. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol Aging 2016;47:35-40. https://doi.org/10.1016/j.neurobiolaging.2016.07.008
- Nuutinen T, Suuronen T, Kyrylenko S, et al. Induction of clusterin/apoJ expression by histone deacetylase inhibitors in neural cells. Neurochem Int 2005;647:528-538. https://doi.org/10.1016/j.neuint.2005.07.007
- Silva PNO, Gigek CO, Leal MF, et al. Promoter methylation analysis of SIRT3, SMARCA5, HTERT and CDH1 genes in aging and Alzheimer’s disease. J Alzheimers Dis 2008;13:173-176. https://doi.org/10.3233/jad-2008-13207
- Smith AR, Smith RG, Condliffe D, et al. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol Aging 2016;47:35-40. https://doi.org/10.1016/j.neurobiolaging.2016.07.008
- Smith RG, Pishva E, Shireby G, et al. A meta-analysis of epigenome-wide association studies in Alzheimer's disease highlights novel differentially methylated loci across cortex. Nat Commun 2021; 12:3517. https://doi.org/10.1038/s41467-021-23243-4
- Furuya TK, Silva PNO, Payão SLM, et al. Analysis of SNAP25 mRNA expression and promoter DNA methylation in brain areas of Alzheimer’s disease patients. Neuroscience 2012;220:41-46. https://doi.org/10.1016/j.neuroscience.2012.06.035
- Furuya TK, Da Silva PNO, Payão SLM, et al. SORL1 and SIRT1 mRNA expression and promoter methylation levels in aging and Alzheimer’s disease. Neurochem Int 2012;61:973-975. https://doi.org/10.1016/j.neuint.2012.07.014
- Sanchez-Mut JV, Aso E, Heyn H, et al. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer's disease. Hippocampus 2014;24:363-368. https://doi.org/10.1002/hipo.22245
- De Jager PL, Srivastava G, Burgess J, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 2014 ;17:1156-1163. https://doi.org/10.1038/nn.3786
- Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci 2014;17:1164–1170. https://doi.org/10.1038/nn.3782
- Shinagawa S, Kobayashi N, Nagata T, et al. DNA methylation in the NCAPH2/LMF2 promoter region is associated with hippocampal atrophy in Alzheimer’s disease and amnesic mild cognitive impairment patients. Neurosci Lett 2016;629:33–37. https://doi.org/10.1016/j.neulet.2016.06.055
- Buira SP, Albasanz JL, Dentesano G, et al. DNA methylation regulates adenosine A(2A) receptor cell surface expression levels. J Neurochem 2010;112:1273-1285. https://doi.org/10.1111/j.1471-4159.2009.06538.x
- Buira SP, Dentesano G, Albasanz JL, et al. DNA methylation and Yin Yang-1 repress adenosine A2A receptor levels in human brain. J Neurochem 2010;115:283-295. https://doi.org/10.1111/j.1471-4159.2010.06928.x
- Villar-Menéndez I, Nuñez F, Díaz-Sánchez S, et al. Striatal adenosine A2A receptor expression is controlled by S-adenosyl-L-methionine-mediated methylation. Purinergic Signal 2014;10:523-528. https://doi.org/10.1007/s11302-014-9417-4
- Piaceri I, Raspanti B, Tedde A, et al. Epigenetic modifications in Alzheimer’s disease: cause or effect? J Alzheimers Dis 2015;43:1169-1173. https://doi.org/10.3233/JAD-141452
- Blanch M, Mosquera JL, Ansoleaga B, et al. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol 2016;186:385-397. https://doi.org/10.1016/j.ajpath.2015.10.004
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet 2001;2:919-929. https://doi.org/10.1038/35103511
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004;5:522-531. https://doi.org/10.1038/nrg1379
- Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell 2009;136: 215-233. https://doi.org/10.1016/j.cell.2009.01.002
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Biol 2009;10:126-139. https://doi.org/10.1038/nrm2632
- Wahid F, Shezhad A, Khan T, et al. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Bioch Biophys Acta - Mol Cell Res 2010;1803:1231-1243. https://doi.org/10.1016/j.bbamcr.2010.06.013
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15-20. https://doi.org/10.1016/j.cell.2004.12.035
- Lewis BP, Shih IH, Jones-Rhoades MW, et al. Prediction of mammalian microRNA targets. Cell 2003;115: 787-798. https://doi.org/10.1016/s0092-8674(03)01018-3
- Boudreau RL, Jiang P, Gilmore BL, et al. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 2014;81: 294-305. https://doi.org/10.1016/j.neuron.2013.10.062
- Tan L, Yu JT, Hu N, et al. Non-coding RNAs in Alzheimer’s disease. Mol Neurobiol 2013;47:382-393. https://doi.org/10.1007/s12035-012-8359-5
- Dehgani R, Rahmani F, Rezaei N. MicroRNA in Alzheimer’s disease revisited: implications for major neuropathological mechanisms. Rev Neurosci 2018;29:161-182. https://doi.org/10.1515/revneuro-2017-0042
- Wang M, Qin L, Tang B. MicroRNAs in Alzheimer’s disease. Front Genet 2019;10: 153. https://doi.org/10.3389/fgene.2019.00153
- Lee ST, Chu K, Jung KH, et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann Neurol 2012;72:269-277. https://doi.org/10.1002/ana.23588
- Hébert SS, Horre K, Nicolai L, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis 2009;33:422-428. https://doi.org/10.1016/j.nbd.2008.11.009
- Hébert SS, Horré K, Nicolai L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc Natl Acad Sci USA 2008;105, 6415-6420. https://doi.org/10.1073/pnas.0710263105
- Patel N, Hoang D, Miller N, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener 2008; 3:10. https://doi.org/10.1186/1750-1326-3-10
- Jarvis CI, Goncalves MB, Clarke E, et al. Retinoic acid receptor-alpha signalling antagonizes both intracellular and extracellular amyloid-beta production and prevents neuronal cell death caused by amyloid-beta. Eur J Neurosci 2010;32:1246-1255. https://doi.org/10.1111/j.1460-9568.2010.07426.x
- Vilardo E, Barbato C, Ciotti M, et al. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem 2010; 285: 18344-18351. https://doi.org/10.1074/jbc.M110.112664
- Liu CG, Wang JL, Li L, et al. MicroRNA-384 regulates both amyloid precursor protein and beta-secretase expression and is a potential biomarker for Alzheimer’s disease. Int J Mol Med 2014;34:160-166. https://doi.org/10.3892/ijmm.2014.1780
- Liu CG, Wang JL, Li L, et al. MicroRNA-135a and -200b, potential biomarkers for Alzheimers disease, regulate beta secretase and amyloid precursor protein. Brain Res 2014;1583: 55-64. https://doi.org/10.1016/j.brainres.2014.04.026
- Liang C, Zhu H, Xu Y, et al. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res 2012;1455: 103-113. https://doi.org/10.1016/j.brainres.2011.10.051
- Long JM, Ray B, Lahiri DK. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J Biol Chem 2012;287: 31298-31310. https://doi.org/10.1074/jbc.M112.366336
- Long JM, Ray B, Lahiri DK. MicroRNA-339-5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem 2014; 289: 5184-5198. https://doi.org/10.1074/jbc.M113.518241
- Cheng C, Li W, Zhang Z, et al. MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10). J Biol Chem 2013;288:13748-13761. https://doi.org/10.1074/jbc.M112.381392
- Deng Y, Ding Y, Hou D. Research status of the regulation of miRNA on BACE1. Int J Neurosci 2014;124:474-477. https://doi.org/10.3109/00207454.2013.858249
- Lei X, Lei L, Zhang Z, et al. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int J Clin Exp Pathol 2015;8:1565-1574. PMID: 25973041.
- Li Q, Li X, Wang L, et al. miR-98-5p acts as a target for Alzheimer’s disease by regulating Aβ production through modulating SNX6 expression. J Mol Neurosci 2016; 60:413-420. https://doi.org/10.1007/s12031-016-0815-7
- Li W, Li X, Xin X, et al. MicroRNA-613 regulates the expression of brain-derived neurotrophic factor in Alzheimer’s disease. Biosci Trends 2016;10: 372-377. https://doi.org/10.5582/bst.2016.01127
- Kim J, Yoon H, Chung DE, et al. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem 2016;137: 436-445. https://doi.org/10.1111/jnc.13507
- An F, Gong G, Wang Y, et al. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017;8:114065-114071. https://doi.org/10.18632/oncotarget.23119
- Gong G, An F, Wang Y et al. miR-15b represses BACE1 expression in sporadic Alzheimer's disease. Oncotarget 2017;8:91551-91557. https://doi.org/10.18632/oncotarget.21177
- Chopra N, Wang R, Maloney B, et al. MicroRNA-298 reduces levels of human amyloid-beta precursor protein (APP), beta-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol Psychiatry 2020;26: 5636-5657. https://doi.org/10.1038/s41380-019-0610-2
- Zhu HC, Wang LM, Wang M, et al. MicroRNA-195 downregulates Alzheimer’s disease amyloid-beta production by targeting BACE1. Brain Res Bull 2012;88:596-601. https://doi.org/10.1016/j.brainresbull.2012.05.018
- Zhu QB, Unmehopa U, Bossers K, et al. MicroRNA-132 and early growth response-1 in nucleus basalis of Meynert during the course of Alzheimer’s disease. Brain 2016;139: 908-921. https://doi.org/10.1093/brain/awv383
- Hu YK, Wang X, Li L, et al. MicroRNA-98 induces an Alzheimer’s disease-like disturbance by targeting insulin-like growth factor 1. Neurosci Bull 2013;29: 745-751. https://doi.org/10.1007/s12264-013-1348-5
- Absalon S, Kochanek DM, Raghavan V, et al. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J Neurosci 2013;33:14645-14659. https://doi.org/10.1523/JNEUROSCI.1327-13.2013
- Dickson JR, Kruse C, Montagna DR, et al. Alternative polyadenylation and miR-34 family members regulate tau expression. J Neurochem 2013;127:739-749. https://doi.org/10.1111/jnc.12437
- Gong J, Zhang J, Li B, et al. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene 2013;32:3071-3079. https://doi.org/10.1038/onc.2012.318
- Zhao Y, Bhattacharjee S, Jones BM, et al. Regulation of neurotropic signaling by the inducible, NF-κB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol 2014; 50: 97-106. https://doi.org/10.1007/s12035-013-8595-3
- Zhang X, Tan L, Lu Y, et al. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett 2015;589:726-729. https://doi.org/10.1016/j.febslet.2015.02.001
- Salta E, Sierksma A, Vanden Eynden E, et al. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med 2016;8:1005-1018. https://doi.org/10.15252/emmm.201606520
- Edbauer D, Neilson JR, Foster KA, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010;65: 373-384. https://doi.org/10.1016/j.neuron.2010.01.005
- Cohen JE, Lee PR, Chen S, et al. MicroRNA regulation of homeostatic synaptic plasticity. Proc Natl Acad Sci USA 2011;108:11650-11655. https://doi.org/10.1073/pnas.1017576108
- Giusti SA, Vogl AM, Brockmann MM, et al. MicroRNA-9 controls dendritic development by targeting REST. Elife 2014:3:e02755. https://doi.org/10.7554/eLife.02755
- Hu Z, Zhao J, Hu T, et al. miR-501-3p mediates the activity-dependent regulation of the expression of AMPA receptor subunit GluA1. J Cell Biol 2015;208:949-959. https://doi.org/10.1083/jcb.201404092
- Olde Loohuis NF, Ba W, Stoerchel PH, et al. MicroRNA-137 Controls AMPA-receptor-mediated transmission and mGluR-dependent LTD. Cell Rep 2015;11:1876-1884. https://doi.org/10.1016/j.celrep.2015.05.040
- Sim SE, Lim CS, Kim JI, et al. The brain-enriched microRNA miR-9-3p regulates synaptic plasticity and memory. J Neurosci 2016;36:8641-8652. https://doi.org/10.1523/JNEUROSCI.0630-16.2016
- Sierksma A, Lu A, Salta E, et al. Deregulation of neuronal miRNAs induced by amyloid-beta or tau pathology. Mol Neurodegener 2018;13:54. https://doi.org/10.1186/s13024-018-0285-1
- Croce N, Gelfo F, Ciotti MT, et al. NPY modulates miR-30a-5p and BDNF in opposite direction in an in vitro model of Alzheimer disease: a possible role in neuroprotection? Mol Cell Biochem 2013;376: 189-195. https://doi.org/10.1007/s11010-013-1567-0
- Shaked I, Meerson A, Wolf Y, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 2009;31: 965-973. https://doi.org/10.1016/j.immuni.2009.09.019
- Shaltiel G, Hanan M, Wolf Y, et al. Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct Funct 2013;218: 59-72. https://doi.org/10.1007/s00429-011-0376-z
- Vo N, Klein ME, Varlamova O, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci USA 2005;102:16426-16431. https://doi.org/10.1073/pnas.0508448102
- Llorens F, Thüne K, Andrés-Benito P, et al. MicroRNA expression in the locus ceruleus, entorhinal cortex, and hippocampus at early and middle stages of Braak neurofibrillary tangle pathology. J Mol Neurosci 2017;63:206-215. https://doi.org/10.1007/s12031-017-0971-4
- Sun C, Jia N, Li R, Zhang Z, et al. miR-143-3p inhibition promotes neuronal survival in an Alzheimer's disease cell model by targeting neuregulin-1. Folia Neuropathol 2020;58:10-21. https://doi.org/10.5114/fn.2020.94002
- Hammad S, Mabondzo A, Hamoudi R, et al. Regulation of P-glycoprotein by miR-27a-3p at the brain endothelial barrier. J Pharm Sci 2022;111:1470-1479. https://doi.org/10.1016/j.xphs.2021.10.021
- Harati R, Hammad S, Tlili A, et al. miR-27a-3p regulates expression of intercellular junctions at the brain endothelium and controls the endothelial barrier permeability. PLoS One 2022;17:e0262152. https://doi.org/10.1371/journal.pone.0262152
- Sochocka M, Zwolińska K, Leszek J. The infectious etiology of Alzheimer's disease. Curr Neuropharmacol. 2017;15:996-1009. https://doi.org/10.2174/1570159X15666170313122937
- Vigasova D, Nemergut M, Liskova B, et al. Multi-pathogen infections and Alzheimer's disease. Microb Cell Fact 2021;20:25. https://doi.org/10.1186/s12934-021-01520-7
- Licastro F, Carbone I, Raschi E, et al. The 21st century epidemic: infections as inductors of neurodegeneration associated with Alzheimer’s disease. Immun Ageing 2014;11:22. https://doi.org/10.1186/1742-4933-11-8
- Maheshwari P, Eslick GD. Bacterial infection and Alzheimer’s disease: a meta-analysis. J Alzheimers Dis 2015;43:957-966. https://doi.org/10.3233/JAD-140621
- Ashraf GM, Tarasov VV, Makhmutovа A, et al. The possibility of an infectious etiology of Alzheimer disease. Mol Neurobiol 2019;56:4479-4491. https://doi.org/10.1007/s12035-018-1388-y
- MacDonald AB, Miranda JM. Concurrent neocortical borreliosis and Alzheimer’s disease. Hum Pathol 1987;18:759-761. https://doi.org/10.1016/s0046-8177(87)80252-6
- Marques AR, Weir SC, Fahle GA, et al. Lack of evidence of Borrelia involvement in Alzheimer's disease. J Infect Dis 2000;182:1006-1007. https://doi.org/10.1086/315792
- Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17:113-118. https://doi.org/10.1046/j.0902-0055.2001.00100.x
- Miklossy J. Alzheimer’s disease: a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinfammation 2011;8:90. https://doi.org/10.1186/1742-2094-8-90
- Miklossy J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 2015;7:46. https://doi.org/10.3389/fnagi.2015.00046
- Senejania AG, Maghsoudloua J, El-Zohirya D, et al. Borrelia burgdorferi co-localizing with amyloid markers in Alzheimer’s disease brain tissues. J Alzheimers Dis 2022;85: 889-903. https://doi.org/10.3233/JAD-215398
- Emery DC, Shoemark DK, Batstone TE, et al. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci. 2017;9:195. https://doi.org/10.3389/fnagi.2017.00195
- Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 2010;5:e9505. https://doi.org/10.1371/journal.pone.0009505
- Kamer AR, Dasanayake AP, Craig RG, et al. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis 2008;13:437-449. https://doi.org/10.3233/jad-2008-13408
- Sparks Stein P, Stefen MJ, Smith C, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement 2012;8:196-203. https://doi.org/10.1016/j.jalz.2011.04.006
- Beydoun MA, Beydoun HA, Weiss J, et al. Helicobacter pylori, periodontal pathogens, and their interactive association with incident all-cause and Alzheimer’s disease dementia in a large national survey. Mol Psychiatry 2021;26:6038-6053. https://doi.org/10.1038/s41380-020-0736-2
- Kamer AR, Craig RG, Pirraglia E, et al. TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Neuroimmunol. 2009;216:92-97. https://doi.org/10.1016/j.jneuroim.2009.08.013
- Abbayya K, Puthanakar NY, Naduwinmani S, et al. Association between periodontitis and Alzheimer’s Disease. J Neuroimmunol 2009;216:92-7.doi: 10.1016/j.jneuroim.2009.08.013.
- Ide M, Harris M, Stevens A, et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One 2016;11:e0151081. https://doi.org/10.1016/j.jneuroim.2009.08.013
- Alonso R, Pisa D, Rabano A, et al. Alzheimer’s disease and disseminated mycoses. Eur J Clin Microbiol Infect Dis 2014b;33:1125-1132. https://doi.org/10.1007/s10096-013-2045-z
- Pisa D, Alonso R, Rábano A, et al. Different brain regions are infected with fungi in Alzheimer's disease. Sci Rep 2015;5:15015. https://doi.org/10.1038/srep15015
- Pisa D, Alonso R, Fernández-Fernández AM, et al. Polymicrobial infections in brain tissuie from Alzheimer’s disease patients. Sci Rep 2017;7:5559. https://doi.org/10.1038/s41598-017-05903-y
- Alonso R, Pisa D, Marina A, et al. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis 2014;41:301-311. https://doi.org/10.3233/JAD-132681
- Alonso R, Pisa D, Marina A, et al. Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int J Biol Sci 2015;11:546-558. https://doi.org/10.7150/ijbs.11084
- Alonso R, Pisa D, Aguado B, et al. Identification of fungal species in brain tissue from Alzheimer’s disease by next-generation sequencing. J Alzheimers Dis 2017;58: 55-67. https://doi.org/10.3233/JAD-170058
- Pisa D, Alonso R, Marina AI, et al. Human and microbial proteins from corpora amylacea of Alzheimer's disease. Sci Rep 2018;8:9880. https://doi.org/10.1038/s41598-018-28231-1
- Alonso R, Pisa D, Fernández-Fernández AM, et al. Fungal infection in neural tissue of patients with amyotrophic lateral sclerosis. Neurobiol Dis 2017;108:249-260. https://doi.org/10.1016/j.nbd.2017.09.001
- Alonso R, Pisa D, Rábano A, et al. Cerebrospinal fuid from Alzheimer’s disease patients contains fungal proteins and DNA. J Alzheimers Dis 2015;47:873-876. https://doi.org/10.3233/JAD-150382
- Ball MJ. Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can J Neurol Sci J 1982;9:303-306. https://doi.org/10.1017/s0317167100044115
- Gannicliffe A, Sutton RN, Itzhaki RF. Viruses, brain and immunosuppression. Psychol Med 1986;16:247-249. https://doi.org/10.1017/s0033291700009053
- Lin W-R, Wozniak MA, Cooper RJ, et al. Herpesviruses in brain and Alzheimer’s disease. J Pathol 2002;197:395-402. https://doi.org/10.1002/path.1127
- Hemling N, Röyttä M, Rinne J, et al. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann Neurol 2003; 54: 267-271. https://doi.org/10.1002/ana.10662
- Warren-Gash C, Forbes HJ, Williamson E, et al. Human herpesvirus infections and dementia or mild cognitive impairment: A systematic review and meta-analysis. Sci Rep 2019;9:4743. https://doi.org/10.1038/s41598-019-41218-w
- Jamieson GA, Maitland NJ, Wilcock GK, et al. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol 1991;33:224-227. https://doi.org/10.1002/jmv.1890330403
- Itzhaki RF, Lin WR, Shang D, et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997;349:241-244. https://doi.org/10.1016/S0140-6736(96)10149-5
- Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J Alzheimers Dis 2015;48:319-353. https://doi.org/10.3233/JAD-142853
- Lövheim H, Gilthorpe J, Johansson A, et al. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimers Dement 2015;11:587-592. https://doi.org/10.1016/j.jalz.2014.07.157
- Itzhaki RF. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front Aging Neurosci 2018;10:324. https://doi.org/10.3389/fnagi.2018.00324
- Wozniak M, Mee A, Itzhaki R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 2009;217:131-138. https://doi.org/10.1002/path.2449
- Lai SW, Kuo YH, Liao KF. Herpes zoster and Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 2021 Jul 23. https://doi.org/10.1007/s00406-021-01311-6
- Barnes LL, Capuano AW, Aiello AE, et al. Cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals. J Infect Dis 2015;211:230-237. https://doi.org/10.1093/infdis/jiu437
- Fotheringham J, Akhyani N, Vortmeyer A, et al. Detection of active human herpesvirus–6 infection in the brain: correlation with polymerase chain reaction detection in cerebrospinal fluid. J Infect Dis 2007;195:450-454. https://doi.org/10.1086/510757
- Readhead B, Haure-Mirande JV, Funk CC, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018;99:64-82.e7. https://doi.org/10.1016/j.neuron.2018.05.023
- Allnutt MA, Johnson K, Bennett DA, et al. Human herpesvirus 6 detection in Alzheimer’s disease cases and controls across multiple cohorts. Neuron 2000; 105:1027-1035. https://doi.org/10.1016/j.neuron.2019.12.031
- Santpere G, Telford M, Andrés-Benito P, et al. The presence of human herpesvirus 6 in the brain in health and disease. Biomolecules 2020;10:1520. https://doi.org/10.3390/biom10111520
- Rodriguez JD, Royall D, Daum LT, et al. Amplification of herpes simplex type 1 and human herpes type 5 viral DNA from formalin-fxed Alzheimer brain tissue. Neurosci Lett 2005;390:37-41. https://doi.org/10.1016/j.neulet.2005.07.052
- Carbone I, Lazzarotto T, Ianni M, et al. Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol Aging 2014;35:122-129. https://doi.org/10.1016/j.neurobiolaging.2013.06.024
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018;99:56-63.e3. https://doi.org/10.1016/j.neuron.2018.06.030
- Ganz T, Fainstein N, Elad A, et al. Microbial pathogens induce neurodegeneration in Alzheimer's disease mice: protection by microglial regulation. J Neuroinflammation 2022;19:5. https://doi.org/10.1186/s12974-021-02369-8
- Phuna ZX, Madhavan P. A reappraisal on amyloid cascade hypothesis: the role of chronic infection in Alzheimer's disease. Int J Neurosci 2022 Mar 14;1-19. https://doi.org/10.1080/00207454.2022.2045290
- Jiang C, Li G, Huang P, et al. The gut microbiota and Alzheimer's disease. J Alzheimers Dis 2017;58:1-15. https://doi.org/10.3233/JAD-161141
- Szablewski L. Human gut microbiota in health and Alzheimer's disease. J Alzheimers Dis 2018;62:549-560. https://doi.org/10.3233/JAD-170908
- Megur A, Baltriukienė D, Bukelskienė V, et al. The microbiota-gut-brain axis and Alzheimer's disease: neuroinflammation is to blame? Nutrients 2020;13:37. https://doi.org/10.3390/nu13010037
- Pluta R, Ułamek-Kozioł M, Januszewski S, et al. Gut microbiota and pro/prebiotics in Alzheimer's disease. Aging 2020;12:5539-5550. https://doi.org/10.18632/aging.102930
- Kesika P, Suganthy N, Sivamaruthi BS, et al. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer's disease. Life Sci 2021;264:118627. https://doi.org/10.1016/j.lfs.2020.118627
- De la Fuente M. The role of the microbiota-gut-brain axis in the health and illness condition: A focus on Alzheimer's disease. J Alzheimers Dis 2021;81:1345-1360. https://doi.org/10.3233/JAD-201587
- Łuc M, Misiak B, Pawłowski M, et al. Gut microbiota in dementia. Critical review of novel findings and their potential application. Prog Neuropsychopharmacol. Biol Psychiatry 2021;104: 110039. https://doi.org/10.1016/j.pnpbp.2020.110039
- Aaldijk E, Vermeiren Y. The role of serotonin within the microbiota-gut-brain axis in the development of Alzheimer's disease: a narrative review. Ageing Res Rev 2022;3: 101556. https://doi.org/10.1016/j.arr.2021.101556
- Rueda-Ruzafa L, Cruz F, Cardona D, et al. Opioid system influences gut-brain axis: Dysbiosis and related alterations. Pharmacol Res 2020;159:104928. https://doi.org/10.1016/j.phrs.2020.104928
- Leaston J, Kulkarni P, Gharagouzloo C, et al. Do we swallow the waste from our brain? Front Neurosci 2021; 15:763780. https://doi.org/10.3389/fnins.2021.763780
- Walker LC, Callahan MJ, Bian F, et al. Exogenous induction of cerebral beta-amyloidosis in betaAPP-transgenic mice. Peptides 2002;23: 1241-1247.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral b-amyloidogenesis is governed by agent and host. Science 2006;313:1781-1784. https://doi.org/10.1126/science.1131864
- Walker LC, Levine H, Mattson MP, et al. Inducible proteopathies. Trends Neurosci 2006;29:438-443. https://doi.org/10.1016/j.tins.2006.06.010
- Rosen RF, Fritz JJ, Dooyema J, et al. Exogenous seeding of cerebral b-amyloid deposition in bAPP-transgenic rats. J Neurochem 2012;120:660-666. https://doi.org/10.1111/j.1471-4159.2011.07551.x
- Morales R, Duran-Aniotz C, Castilla J, et al. De novo induction of amyloid-b deposition in vivo. Mol Psychiatry 2012;17:1347-1353. https://doi.org/10.1038/mp.2011.120
- Langer F, Eisele YS, Fritschi SK, et al. Soluble Aβ seeds are potent inducers of cerebral b-amyloid deposition. J Neurosci 2011;31: 14488-14495. https://doi.org/10.1523/JNEUROSCI.3088-11.2011
- Hamaguchi T, Eisele Y, Varvel N, et al. The presence of Ab seeds, and not age per se, is critical to the initiation of Ab deposition in the brain. Acta Neuropathol 2012;123:31-37. https://doi.org/10.1007/s00401-011-0912-1
- Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 2011;14:750-756. https://doi.org/10.1038/nn.2801
- Eisele YS, Obermüller U, Heilbronner G et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010;330:980-982. https://doi.org/10.1126/science.1194516
- Forny-Germano L, Lyra de Silva NM, Batista AF, et al. Alzheimer's disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J Neurosci 2014;34:13629-13643. https://doi.org/10.1523/JNEUROSCI.1353-14.2014
- Lauwers E, Lalli G, Brandner S, et al. Potential human transmission of amyloid-beta pathology: surveillance and risks. Lancet Neurol 2020;19:872-878. https://doi.org/10.1016/S1474-4422(20)30238-6
- Pooler AM, Phillips EC, Lau DHW, et al. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 2013;14:389-394. https://doi.org/10.1038/embor.2013.15
- Yamada K, Holth JK, Liao F, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med 2014; 211:387-393. https://doi.org/10.1084/jem.2013168
- Wu JW, Hussaini SA, Bastille IM, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 2016;19:1085-1092. https://doi.org/10.1038/nn.4328
- Schultz MK, Gentzel R, Usenovic M, et al. Pharmacogenetic neuronal stimulation increases human tau pathology and trans-synaptic spread of tau to distal brain regions in mice. Neurobiol Dis 2018;118:161-176. https://doi.org/10.1016/j.nbd.2018.07.003
- Saman S, Kim WH, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 2012;287:3842-3849. https://doi.org/10.1074/jbc.M111.277061
- Takeda S, Wegmann S, Cho H, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun 2015;6: 8490. https://doi.org/10.1038/ncomms9490
- Mudner A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 2017; 5:99. https://doi.org/10.1186/s40478-017-0488-7
- Dujardin S, Hyman BT. Tau prion-like propagation: state of the art and current challenges. Adv Exp Med Biol 2019;1184:305-325. https://doi.org/10.1007/978-981-32-9358-8_23
- Goedert M. Tau proteinopathies and the prion concept. Prog Mol Biol Transl Sci 2020;175: 239-259. https://doi.org/10.1016/bs.pmbts.2020.08.003
- Peng C, Trojanowski JQ, Lee VMY. Protein transmission in neurodegenerative disease. Nat Rev Neurol 2020;16:199-212. https://doi.org/10.1038/s41582-020-0333-7
- de Calignon A, Polydoro M, Suárez-Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 2012;73:685-697. https://doi.org/10.1016/j.neuron.2011.11.033
- Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7:e31302. https://doi.org/10.1371/journal.pone.0031302
- Dujardin S, Lécolle K, Caillierez R, et al. Neuron-to-neuron wild-type tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun 2014; 2:14. https://doi.org/10.1186/2051-5960-2-14
- Fauré J, Lachenal G, Court M, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 2006;31, 642-648. https://doi.org/10.1016/j.mcn.2005.12.003
- Smalheiser NR. Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol Direct 2007;2:35. https://doi.org/10.1186/1745-6150-2-35
- Frost B, Jacks RL, Diamond MI. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 2009; 284:12845-12852. https://doi.org/10.1074/jbc.M808759200
- Simons M, Raposo G. Exosomes--vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575-581. https://doi.org/10.1016/j.ceb.2009.03.007
- Wu JW, Herman M, Liu L, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 2013; 288:1856-1870. https://doi.org/10.1074/jbc.M112.394528
- Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA 2013;110:E3138-E3147. https://doi.org/10.1073/pnas.1301440110
- Saman S, Lee NC, Inoyo I, et al. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer's disease. J Alzheimers Dis 2014;40:S47-70. https://doi.org/10.3233/JAD-132135
- Polanco JC, Scicluna BC, Hill AF, et al. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem 2016;291:12445-12466. https://doi.org/10.1074/jbc.M115.709485
- Wang Y, Balaji V, Kaniyappan S, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 2017;12: 5. https://doi.org/10.1186/s13024-016-0143-y
- Demaegd K, Schymkowitz J, Rousseau F. Transcellular spreading of tau in tauopathies. Chembiochem 2018;19:2424-2432. https://doi.org/10.1002/cbic.201800288
- Jiang L, Dong H, Cao H, et al. Exosomes in pathogenesis, diagnosis, and treatment of Alzheimer's disease. Med Sci Monit 2019;25:3329-3335. https://doi.org/10.12659/MSM.914027
- Pérez M, Avila J, Hernández F. Propagation of tau via extracellular vesicles. Front Neurosci 2019;13:698. https://doi.org/10.3389/fnins.2019.00698
- Iba M, Guo JL, McBride JD, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci 2013; 33: 1024-1037. https://doi.org/10.1523/JNEUROSCI.2642-12.2013
- Peeraer E, Bottelbergs A, Van Kolen K, et al. Intracerebral injection of preformed synthetic fibrils initiates widesprad tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 2015;73:83-95. https://doi.org/10.1016/j.nbd.2014.08.032
- Ahmed Z, Cooper J, Murray TK, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 2014;127:667-683. https://doi.org/10.1007/s00401-014-1254-6
- Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 111:909-913. https://doi.org/10.1038/ncb1901
- Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013; 110:9535-9540. https://doi.org/10.1073/pnas.1301175110
- Boluda S, Iba M, Zhang B, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 2015;129:221–237. https://doi.org/10.1007/s00401-014-1373-0
- Hu W, Zhang X, Tung YC, et al. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement 2016;12:1066-1077. https://doi.org/10.1016/j.jalz.2016.01.014
- Audouard E, Houben S, Masaracchia C, et al. High-molecular weight paired helical filaments from Alzheimer brain induces seeding of wild-type mouse tau into argyrophilic 4R tau pathology in vivo. Am J Pathol 2016; 186:2709-2722. https://doi.org/10.1016/j.ajpath.2016.06.008
- Guo JL, Narasimhan S, Changolkar L, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 2016; 213:2635–2654. https://doi.org/10.1084/jem.20160833
- Narashima S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in non-transgenic mouse brain. J Neurosci 2017;37: 11406-11423. https://doi.org/10.1523/JNEUROSCI.1230-17.2017
- Ferrer I, Aguiló García M, López González I, et al. Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol 2018; 28: 965-985. https://doi.org/10.1111/bpa.12593
- Ferrer I, Andrés-Benito P, Sala-Jarque J, et al. Capacity for seeding and spreading of argyrophilic grain disease in a wild-type murine model; comparisons with primary age-related tauopathy. Front Mol Neurosci 2020; 13:101. https://doi.org/10.3389/fnmol.2020.00101
- Ferrer I, Andrés-Benito P, Zelaya MV, et al. Familial globular glial tauopathy linked to MAPT mutations: molecular neuropathology and seeding capacity of a prototypical mixed neuronal and glial tauopathy. Acta Neuropathol 2020;139:735-771. https://doi.org/10.1007/s00401-019-02122-9
- Ferrer I, Aguiló García M, Carmona M, et al. Involvement of oligodendrocytes in tau seeding and spreading in tauopathies. Front Aging Neurosci 2019; 11:112. https://doi.org/10.3389/fnagi.2019.00112
- Weitzman SA, Narasimhan S, He Z, et al. Insoluble tau from human FTDP-17 cases exhibit unique transmission properties in vivo. J Neuropathol Exp Neurol 2020;79: 941-949. https://doi.org/10.1093/jnen/nlaa086
- Clavaguera F, Hench J, Goedert M, et al. Invited review: prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 2015;41:47-58. https://doi.org/10.1111/nan.12197
- Goedert M, Spillantini MG. Propagation of tau aggregates. Mol Brain 2017; 10:18. https://doi.org/10.1186/s13041-017-0298-7
- Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 2017; 40:189-210. https://doi.org/10.1146/annurev-neuro-072116-031153
- Vaquer-Alicea J, Diamond MI, Joachimiak LA. Tau strains shape disease. Acta Neuropathologica 2021;142:57-71. https://doi.org/10.1007/s00401-021-02301-7
- Li L, Shi R, Gu J, et al. Alzheimer's disease brain contains tau fractions with differential prion-like activities. Acta Neuropathol Commun 2021;9:28. https://doi.org/10.1186/s40478-021-01127-4
- Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med 2020;26:1256-1263. https://doi.org/10.1038/s41591-020-0938-9
- Janke C, Beck M, Sthal T, et al. Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Brain Res Mol Brain Res 1999;68:119-128. https://doi.org/10.1016/s0169-328x(99)00079-0
- Ferrer I, Zelaya V, Aguio M, et al. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol 2020;30:298-318. https://doi.org/10.1111/bpa.12778
- Beckman D, Chakrabarty P, Ott S, et al. A novel tau-based rhesus monkey model of Alzheimer's pathogenesis. Alzheimers Dement 2021;17:933-945. https://doi.org/10.1002/alz.12318
- Clavaguera F, Hench J, Lavenir I, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 2014; 127: 299-301. https://doi.org/10.1007/s00401-013-1231-5
- Furman JL, Vaquer-Alicea J, White CL, et al. Widespread tau seeding activity at early Braak stages. Acta Neuropathol 2017;133: 91-100. https://doi.org/10.1007/s00401-016-1644-z
- Kaufman SK, Del Tredici K, Thomas TL, et al. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and part. Acta Neuropathol 2018; 136:57-67. https://doi.org/10.1007/s00401-018-1855-6
- Seemiller J, Bischof GN, Hoenig MC, et al. Indication of retrograde tau spreading along Braak stages and functional connectivity pathways. Eur J Nucl Med Mol Imaging 2021; 48: 2272-2282. https://doi.org/10.1007/s00259-020-05183-1
- Meisl G, Hidari E, Allinson K, et al. In vivo rate-determining steps of tau seed accumulation in Alzheimer's disease. Sci Adv 2021;7:eabh1448. https://doi.org/10.1126/sciadv.abh1448
- Andrés-Benito P, Carmona M, Jordán M, et al. Host tau genotype specifically designs and regulates tau seeding and spreading and host tau transformation following intrahippocampal injection of identical tau AD inoculum. Int J Mol Sci 2022;23:718. https://doi.org/10.3390/ijms23020718
- Detrez JR, Maurin H, Van Kolen K, et al. Regional vulnerability and spreading of hyperphosphorylated tau in seeded mouse brain. Neurobiol Dis 2019;127:398-409. https://doi.org/10.1016/j.nbd.2019.03.010
- Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015;138:1738-1755. https://doi.org/10.1093/brain/awv081
- Bolós M, Llorens-Martín M, Jurado-Arjona J, et al. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis 2016;50:77-87. https://doi.org/10.3233/JAD-150704
- Perea JR, Llorens-Martín M, Ávila J, et al. The role of microglia in the spread of tau: relevance for tauopathies. Front Cell Neurosci 2018;12:172. https://doi.org/10.3389/fncel.2018.00172
- Španić E, Langer Horvat L, Hof PR, et al. Role of microglial cells in Alzheimer's disease tau propagation. Front Aging Neurosci 2019;11:271. https://doi.org/10.3389/fnagi.2019.00271
- Brelstaff JH, Mason M, Katsinelos T, et al. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv 2021;7:eabg4980. https://doi.org/10.1126/sciadv.abg4980
- Gratuze M, Chen Y, Parhizkar S, et al. Activated microglia mitigate Aβ-associated tau seeding and spreading. J Exp Med 2021;218:e20210542. https://doi.org/10.1084/jem.20210542
- Hopp SC, Lin Y, Oakley D, et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation 2018;15:269. https://doi.org/10.1186/s12974-018-1309-z
- Jellinger KA, Stadelmann C. Problems of cell death in neurodegeneration and Alzheimer's disease. J Alzheimers Dis 2001;3:31-40. https://doi.org/10.3233/jad-2001-3106
- Roth KA. Caspases, apoptosis, and Alzheimer disease: causation, correlation, and confusion. J Neuropathol Exp Neurol 2001;60:829-838. https://doi.org/10.1093/jnen/60.9.829
- Obulesu M, Lakshmi MJ. Apoptosis in Alzheimer's disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 2014;39:2301-2312. https://doi.org/10.1007/s11064-014-1454-4
- Fasulo L, Ugolini G, Visintin M, et al. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 2000;75:624-633. https://doi.org/10.1046/j.1471-4159.2000.0750624.x
- Chung CW, Song YH, Kim KK, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 2001;8:162-172. https://doi.org/10.1006/nbdi.2000.0335
- Zilkova M, Zilka N, Kovac A, et al. Hyperphosphorylated truncated protein tau induces caspase-3 independent apoptosis-like pathway in the Alzheimer's disease cellular model. Alzheimers Dis 2011;23:161-169. https://doi.org/10.3233/JAD-2010-101434
- Gómez-Ramos A, Díaz-Hernández M, Cuadros R, et al. Extracellular tau is toxic to neuronal cells. FEBS Lett 2006;580:4842-4850. https://doi.org/10.1016/j.febslet.2006.07.078
- Caccamo A, Branca C, Piras IS, et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci 2017;20:1236-1246. https://doi.org/10.1038/nn.4608
- Royce GH, Brown-Borg HM, Deepa SS. The potential role of necroptosis in inflammaging and aging. Geroscience 2019;41:795-811. https://doi.org/10.1007/s11357-019-00131-w
- Dionísio PA, Amaral JD, Rodrigues CMP. Molecular mechanisms of necroptosis and relevance for neurodegenerative diseases. Int Rev Cell Mol Biol 2020;353:31-82. https://doi.org/10.1016/bs.ircmb.2019.12.006
- Gómez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 1997;41:17-24. https://doi.org/10.1002/ana.410410106
- van de Nes JAP, Nafe R, Schlote W. Non-tau based neuronal degeneration in Alzheimer's disease -- an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res 2008;1213:152-165. https://doi.org/10.1016/j.brainres.2008.03.043
- Takahashi RH, Nagao T, Gouras GK. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer's disease. Pathol Int 2017;67:185-193. https://doi.org/10.1111/pin.12520
- Jeong S. Molecular and cellular basis of neurodegeneration in Alzheimer's disease. Mol Cells 2017;40:613-620. https://doi.org/10.14348/molcells.2017.0096
- Leong YQ, Ng KY, Chye SM, et al. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab Brain Dis 2020;35:11-30. https://doi.org/10.1007/s11011-019-00516-y
- Siman-Tov T, Bosak N, Sprecher E, et al. Early Age-Related Functional Connectivity Decline in High-Order Cognitive Networks. Front Aging Neurosci 2017;8:330. https://doi.org/10.3389/fnagi.2016.00330
- Tsvetanov KA, Henson RN, Tyler LK, et al. Extrinsic and intrinsic brain network connectivity maintains cognition across the lifespan despite accelerated decay of regional brain activation. J Neurosci 2016;36:3115-3126. https://doi.org/10.1523/JNEUROSCI.2733-15.2016
- Buckley RF, Schultz AP, Hedden T, et al. Functional network integrity presages cognitive decline in preclinical Alzheimer disease. Neurology 2017;89:29-37. https://doi.org/10.1212/WNL.0000000000004059
- Jockwitz C, Caspers S. Resting-state networks in the course of aging-differential insights from studies across the lifespan vs. amongst the old. Pflugers Arch 2021;473:793-803. https://doi.org/10.1007/s00424-021-02520-7
- Zhang H, Gertel VH, Cosgrove AL, et al. Age-related differences in resting-state and task-based network characteristics and cognition: a lifespan sample. Neurobiol Aging 2021;101:262-272. https://doi.org/10.1016/j.neurobiolaging.2020.10.025
- Hoenig MC, Bischof GN, Seemiller J, et al. Networks of tau distribution in Alzheimer's disease. Brain 2018;141:568-581. https://doi.org/10.1093/brain/awx353
- Zheng W, Yao Z, Xie Y, et al. Identification of Alzheimer's disease and mild cognitive impairment using networks constructed based on multiple morphological brain features. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:887-897. https://doi.org/10.1016/j.bpsc.2018.06.004
- Pelkmans W, Ossenkoppele R, Dicks E, et al. Tau-related grey matter network breakdown across the Alzheimer's disease continuum. Alzheimers Res Ther 2021;13:138. https://doi.org/10.1186/s13195-021-00876-7
- Shigemoto Y, Sone D, Okita K, et al. Gray matter structural networks related to (18)F-THK5351 retention in cognitively normal older adults and Alzheimer's disease patients. Neurological Sci 2021;22:100309. https://doi.org/10.1016/j.ensci.2021.100309
- Smith RX, Strain JF, Tanenbaum A, et al. Resting-state functional connectivity disruption as a pathological biomarker in autosomal dominant Alzheimer disease. Brain Connect 2021;11:239-249. https://doi.org/10.1089/brain.2020.0808
- Wu Z, Gao Y, Potter T, et al. Interactions between aging and Alzheimer’s disease on structural brain networks. Aging Neurosci 2021;13:639795. https://doi.org/10.3389/fnagi.2021.639795
- Hasani SA, Mayeli M, Salehi MA, et al. A systematic review of the association between amyloid-beta and tau pathology with functional connectivity alterations in the Alzheimer dementia spectrum utilizing PET scan and rsfMRI. Dement Geriatr Cogn Dis Extra 2021;11:78-90. https://doi.org/10.1159/000516164
- Ewers M, Luan Y, Frontzkowski L, et al. Segregation of functional networks is associated with cognitive resilience in Alzheimer's disease. Brain 2021;144:2176-2185. https://doi.org/10.1093/brain/awab112
- King-Robson J, Wilson H, Politis M, et al. Associations between amyloid and tau pathology, and connectome alterations, in Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis 2021;82:541-560. https://doi.org/10.3233/JAD-201457
- Cassady KE, Adams JN, Chen X, et al. Alzheimer's pathology is associated with dedifferentiation of intrinsic functional memory networks in aging. Cereb Cortex 2021;31:4781-4793. https://doi.org/10.1093/cercor/bhab122
- Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957-965. https://doi.org/10.1016/S1474-4422(13)70194-7
- Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014;13:614-629. https://doi.org/10.1016/S1474-4422(14)70090-0
- Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: A systematic review and meta-analysis. Lancet Neurol 2016;15:673-684. https://doi.org/10.1016/S1474-4422(16)00070-3
- Sperling RA, Donohue MC, Raman R, et al. Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol 2020;77:735-745. https://doi.org/10.1001/jamaneurol.2020.0387
- Zhang K, Mizuma H, Zhang X, et al. PET imaging of neural activity, beta-amyloid, and tau in normal brain aging. Eur J Nucl Med Mol Imaging 2021;48:3859-3871. https://doi.org/10.1007/s00259-021-05230-5
- Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer's disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 2016;12: 292-323. https://doi.org/10.1016/j.jalz.2016.02.002
- Rossini PM, Di Iorio R, Vecchio F, et al. Early diagnosis of Alzheimer's disease: the role of biomarkers including advanced EEG signal analysis. Report from the IFCN-sponsored panel of experts. Clin Neurophysiol 2020;131:1287-1310. https://doi.org/10.1016/j.jalz.2016.02.002
- Zetterberg H, Bendlin BB. Biomarkers for Alzheimer’s disease-preparing for a new era of disease-modifying therapies. Mol Psychiatry 2021;26:296-308. https://doi.org/10.1038/s41380-020-0721-9
- Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood-bassed biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol 2022;21:66-77. https://doi.org/10.1016/S1474-4422(21)00361-6
- Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 2015;313:1924-1938. https://doi.org/10.1001/jama.2015.4668
- Jansen WJ, Janssen O, Tijms BM, et al. Prevalence estimates of amyloid abnormality across the Alzheimer disease clinical spectrum. JAMA Neurol 2022;79:228-243. https://doi.org/10.1001/jamaneurol.2021.5216
- Okamura N, Harada R, Furumoto S, et al. Tau PET imaging in Alzheimer's disease. Curr Neurol Neurosci Rep 2014;14:500. https://doi.org/10.1007/s11910-014-0500-6
- Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron 2016;89:971-982. https://doi.org/10.1007/s11910-014-0500-6
- Lockhart SN, Baker SL, Okamura N, et al. Dynamic PET measures of tau accumulation in cognitively normal older adults and Alzheimer’s disease patients measured using [18F] THK-5351. PLoS One 2016;11:e0158460. https://doi.org/10.1371/journal.pone.0158460
-
- Maass A, Landau S, Baker SL, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage 2017; 157:448-463. https://doi.org/10.1016/j.neuroimage.2017.05.058
- Chotipanich C, Jantarato A, Kunawudhi A, et al. 11C-Pittsburgh compound B and 18F-THK 5351 positron emission tomography brain imaging in cognitively normal individuals. World J Nucl Med 2020;20:133-138. https://doi.org/10.4103/wjnm.WJNM_57_20
- Mueller A, Bullich S, Barret O, et al. Tau PET imaging with 18F-PI-2620 in patients with Alzheimer disease and healthy controls: A first-in-humans Study. J Nucl Med 2020;61:911-919. https://doi.org/10.2967/jnumed.119.236224
- Sintini I, Whitwell JL. Update on neuroimaging in Alzheimer's disease. Curr Opin Neurol 2021;34:525-531. https://doi.org/10.1097/WCO.0000000000000947
- Ossenkoppele R, Smith R, Mattsson-Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: A head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol 2021;78:961-971. https://doi.org/10.1001/jamaneurol.2021.1858
- Jack CR, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 2017;16:435-444. https://doi.org/10.1016/S1474-4422(17)30077-7
- Lowe VJ, Wiste HJ, Senjem ML, et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 2018;141:271-287. https://doi.org/10.1093/brain/awx320
- Yoon B, Guo T, Provost K, et al. Abnormal tau in amyloid PET negative individuals. Neurobiol Aging 2022;109:125-134. https://doi.org/10.1016/j.neurobiolaging.2021.09.019
- Groot C, Doré V, Robertson J, et al. Mesial temporal tau is related to worse cognitive performance and greater neocortical tau load in amyloid-beta-negative cognitively normal individuals. Neurobiol Aging 2021;97:41-48. https://doi.org/10.1016/j.neurobiolaging.2020.09.017
- Wisse LEM, Xie L, Das SR, et al. Tau pathology mediates age effects on medial temporal lobe structure. Neurobiol Aging 2022;109:135-144. https://doi.org/10.1016/j.neurobiolaging.2021.09.017
- Chen SD, Lu JY, Li HQ, et al. Staging tau pathology with tau PET in Alzheimer's disease: a longitudinal study. Transl Psychiatry 2021;11:483. https://doi.org/10.1038/s41398-021-01602-5
- Rubinski A, Tosun D, Franzmeier N, et al. Lower cerebral perfusion is associated with tau-PET in the entorhinal cortex across the Alzheimer's continuum. Neurobiol Aging 2021;102:111-118. https://doi.org/10.1016/j.neurobiolaging.2021.02.003
- Berron D, Vogel JW, Insel PS, et al. Early stages of tau pathology and its associations with functional connectivity, atrophy and memory. Brain 2021;144:2771-2783. https://doi.org/10.1093/brain/awab114
- Shen XN, Kuo K, Yang YX, et al. Subtle cognitive impairment as a marker of Alzheimer's pathologies and clinical progression in cognitively normal individuals. Alzheimers Dement 2021;13:e12198. https://doi.org/10.1002/dad2.12198
- Sanchez JS, Becker JA, Jacobs HIL, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci Transl Med 2021;13:eabc0655. https://doi.org/10.1126/scitranslmed.abc0655
- Sanchez JS, Hanseeuw BJ, Lopera F, et al. Longitudinal amyloid and tau accumulation in autosomal dominant Alzheimer's disease: findings from the Colombia-Boston (COLBOS) biomarker study. Alzheimers Res Ther 2021;13:27. https://doi.org/10.1186/s13195-020-00765-5
- Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 2009;68:774–784. https://doi.org/10.1097/NEN.0b013e3181aacbe9
- Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755-766. https://doi.org/10.1007/s00401-014-1349-0
- Jellinger KA, Alafuzoff I, Attems J, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015; 129: 757-762. https://doi.org/10.1007/s00401-015-1407-2
- Bell WR, An Y, Kageyama Y, et al. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimers Dement 2019; 15:8-16. https://doi.org/10.1016/j.jalz.2018.07.215
- Teylan M, Mock C, Gauthreaux K, et al. Cognitive trajectory in mild cognitive impairment due to primary age-related tauopathy. Brain 2020; 143; 611-621. https://doi.org/10.1093/brain/awz403
- Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70: 960-969. https://doi.org/10.1097/NEN.0b013e318232a379
- Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia). Brain Pathol 1998; 8: 367-376. https://doi.org/10.1111/j.1750-3639.1998.tb00160.x
- Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 2010: 119:421-433. https://doi.org/10.1007/s00401-010-0654-5
- Jellinger KA. Different patterns of hippocampal tau pathology in Alzheimer's disease and PART. Acta Neuropathol 2018; 136:811-813. https://doi.org/10.1007/s00401-018-1894-z
- Zhang L, Jiang Y, Zhu J, et al. Quantitative assessment of hippocampal tau pathology in AD and PART. J Mol Neurosci 2020;70:1808-1811. https://doi.org/10.1007/s12031-020-01573-0
- Duyckaerts C, Braak H, Brion JP, et al. PART is part of Alzheimer disease. Acta Neuropathol 2015; 129:749-756. https://doi.org/10.1007/s00401-015-1390-7
- McMillan CT, Lee EB, Jefferson-George K, et al. Alzheimer’s genetic risk is reduced in primary age-related tauopathy: a potential model of resistance? Annals of Clin Transl Neurol 2018; 5: 927-934. https://doi.org/10.1002/acn3.581
- Santa-Maria I, Haggiagi A, Liu X, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol 2012;124:693–704. https://doi.org/10.1007/s00401-012-1017-1
- Schmidt C, Wolff M, Weitz M, et al. Rapidly progressive Alzheimer disease. Arch Neurol 2011; 68:1124-1130. https://doi.org/10.1001/archneurol.2011.189
- Stoeck K, Schmitz M, Ebert E, et al. Immune responses in rapidly progressive dementia: a comparative study of neuroinflammatory markers in Creutzfeldt-Jakob disease, Alzheimer’s disease and multiple sclerosis. J Neuroinflammation 2014;11:170. https://doi.org/10.1186/s12974-014-0170-y
- Schmidt C, Haik S, Satoh K, et al. Rapidly progressive Alzheimer’s disease: a multicenter update. J Alzheimer’s Dis 2012;30:751-756. https://doi.org/10.3233/JAD-2012-120007
- Cohen M, Appleby B, Safar JG. Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion 2016;10:9-17. https://doi.org/10.1080/19336896.2015.1123371
- Drummond E, Nayak S, Faustin A, et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol 2017;133:933-954. https://doi.org/10.1007/s00401-017-1691-0
- Zafar S, Shafiq M, Younas N, et al. Prion protein interactome: identifying novel targets in slowly and rapidly progressive forms of Alzheimer's Disease. J Alzheimers Dis 2017;59:265-275. https://doi.org/10.3233/JAD-170237
- Shafiq M, Zafar S, Younas N, et al. Prion protein oligomers cause neuronal cytoskeletal damage in rapidly progressive Alzheimer's disease. Mol Neurodegener 2021;16:11. https://doi.org/10.1186/s13024-021-00422-x
- Noor A, Zafar S, Shafiq M, et al. Molecular profiles of amyloid‑β proteoforms in typical and rapidly progressive Alzheimer’s disease. Mol Neurobiol 2022 59:17-34. https://doi.org/10.1007/s12035-021-02566-9
- Younas N, Zafar S, Shafiq M, et al. SFPQ and Tau: critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol 2020;140:317-339. https://doi.org/10.1007/s00401-020-02178-y
- Arenaza-Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology 2018;90:695-703. https://doi.org/10.1212/WNL.0000000000005303
- Andersen SL. Centenarians as models of resistance and resilience to Alzheimer's disease and related dementias. Adv Geriatr Med Res 2020;2:e200018. https://doi.org/10.20900/agmr20200018
- Beker N, Ganz A, Hulsman M, et al. Association of cognitive function trajectories in centenarians with postmortem neuropathology, physical health, and other risk factors for cognitive decline. JAMA 2021;4:e2031654. https://doi.org/10.1001/jamanetworkopen.2020.31654
- Giannakopoulos P, Hof PR, Surini M, et al. Quantittaive immunohistochemical analysis of the distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of nonagenarians and centenerians. Acta Neuropathol 1993;85:602-610. https://doi.org/10.1007/BF00334669
- Giannakopoulos P, Hof PR, Vallet PG, et al. Quantitative analysis of neuropathologic changes in the cerebral cortex of centenarians. Prog Neuropsychopharmacol Biol Psychiatry 1995;19:577-592. https://doi.org/10.1016/0278-5846(95)00103-3
- Rogalski E, Gefen T, Mao Q, et al. Cognitive trajectories and spectrum of neuropathology in SuperAgers: The first 10 cases. Hippocampus 2019; 29:458-467. https://doi.org/10.1002/hipo.22828
- Xuereb JH, Brayne C, Dufouil C, et al. Neuropathological findings in the very old. Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann N Y Acad Sci 2000;903:490-496. https://doi.org/10.1111/j.1749-6632.2000.tb06404.x
- Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012;11:1006-1012. https://doi.org/10.1016/S1474-4422(12)70191-6
- Lesuis SL, Hoeijmakers L, Korosi A, et al. Vulnerability and resilience to Alzheimer's disease: early life conditions modulate neuropathology and determine cognitive reserve. Alzheimers Res Ther 2018;10:95. https://doi.org/10.1186/s13195-018-0422-7
- Montine TJ, Cholerton BA, Corrada MM, et al. Concepts for brain aging: resistance, resilience, reserve, and compensation. Alzheimers Res Ther 2019;11:22. https://doi.org/10.1186/s13195-019-0479-y
- Weisenbach SL, Kim J, Hammers D, et al. Linking late life depression and Alzheimer's disease: mechanisms and resilience. Curr Behav Neurosci Rep 2019;6:103-112. https://doi.org/10.1007/s40473-019-00180-7
- Peng S, Roth AR, Apostolova LG, et al. Cognitively stimulating environments and cognitive reserve: The case of personal social networks. Neurobiol Aging 2022;112: 197-203. https://doi.org/10.1016/j.neurobiolaging.2022.01.004
- Zhang X, Alshakhshir N, Zhao L. Glycolytic metabolism, brain resilience, and Alzheimer's disease. Front Neurosci 2021;15:662242. https://doi.org/10.3389/fnins.2021.662242
- Camandola S, Plick N, Mattson MP. Impact of coffee and cacao metabolites on neuroplasticity and neurodegenerative disease. Neurochem Res 2019;44:214-227. https://doi.org/10.1007/s11064-018-2492-0
- Aiello Bowles EJ, Crane PK, Walker RL, et al. Cognitive resilience to Alzheimer’s disease pathology in the human brain. J Alzheimers Dis 2019;68:1071-1083. https://doi.org/10.3233/JAD-180942
- Buciuc M, Wennberg AM, Weigand SD, et al. Effect Modifiers of TDP-43-Associated Hippocampal Atrophy Rates in Patients with Alzheimer's Disease Neuropathological Changes. J Alzheimers Dis 2020a;73:1511-1523. https://doi.org/10.3233/JAD-191040
- Buciuc M, Whitwell JL, Tosakulwong N, et al. Association between transactive response DNA-binding protein of 43 kDa type and cognitive resilience to Alzheimer's disease: a case-control study. Neurobiol Aging 2020b;92:92-97. https://doi.org/10.1016/j.neurobiolaging.2020.04.001
- Nygaard HB, Erson-Omay EZ, Wu X, et al. Whole-exome sequencing of an exceptional longevity cohort. J Gerontol A Biol Sci Med Sci 2019;74:1386-1390. https://doi.org/10.1093/gerona/gly098
- Seto M, Weiner RL, Dumitrescu L, et al. Protective genes and pathways in Alzheimer's disease: moving towards precision interventions. Mol Neurodegener 2021; 16:29. https://doi.org/10.1186/s13024-021-00452-5
- McDermott KL, McFall GP, Andrews SJ, et al. Memory resilience to Alzheimer’s genetic risk: sex effects in predictor profiles. J Gerontol B Psychol Sci Soc Sci 2017;72:937-946. https://doi.org/10.1093/geronb/gbw161
- Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 2020;143:2561-2575. https://doi.org/10.1093/brain/awaa209
- Tavana JP, Rosene M, Jensen NO, et al. RAB10 an Alzheimer’s disease resilience locus and potential drug target. Clin Interv Aging 2018; 14: 73-79. https://doi.org/10.2147/CIA.S159148
- Neuner SM, Wilmott LA, Hoffmann BR, et al. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer's disease mouse models. Behav Brain Res 2017;322:288-298. https://doi.org/10.1016/j.bbr.2016.06.002
- Leng K, Li E, Eser R, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat Neurosci 2021;24:276-287. https://doi.org/10.1038/s41593-020-00764-7
- Barker SJ, Raju RM, Milman NEP, et al. MEF2 is a key regulator of cognitive potential and confers resilience to neurodegeneration. Sci Transl Med 2020;13:eabd7695. https://doi.org/10.1126/scitranslmed.abd7695
- Tesi N, van der Lee SJ, Hulsman M, et al. Immune response and endocytosis pathways are associated with the resilience against Alzheimer's disease. Transl Psychiatry 2020;10:332. https://doi.org/10.1038/s41398-020-01018-7
- Perneczky R, Kempermann G, Korczyn AD, et al. Translational research on reserve against neurodegenerative disease: consensus report of the International Conference on Cognitive Reserve in the Dementias and the Alzheimer's Association Reserve, Resilience and Protective Factors Professional Interest Area working groups. BMC Med 2019;17:47. https://doi.org/10.1186/s12916-019-1283-z
- Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 2015;14:712-720. https://doi.org/10.1080/15384101.2015.1004937
- Arendt T, Rödel L, Gärtner U, et al. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport 1996;7:3047-3049. https://doi.org/10.1097/00001756-199611250-00050
- McShea A, Harris PL, Webster KR, et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol 1997;150:1933-1939. PMID: 9176387.
- Nagy Z, Esiri MM, Cato AM, et al. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol 1997;94:6-15. https://doi.org/10.1007/s004010050665
- Smith MZ, Nagy Z, Esiri MM. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci Lett 1999;271:45-48. https://doi.org/10.1016/s0304-3940(99)00509-1
-
- Arendt T, Holzer M, Gärtner U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer's disease. J Neural Transm 1998;105:949-960. https://doi.org/10.1007/s007020050104
- Raina AK, Zhu X, Rottkamp CA, et al. Cyclin toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J Neurosci Res 2000;61:128-133. https://doi.org/10.1002/1097-4547(20000715)61:2<128::AID-JNR2>3.0.CO;2-H
- Arendt T. Alzheimer's disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brain. Neurobiol Aging 2000;21:783-796. https://doi.org/10.1016/s0197-4580(00)00216-5
- Ueberham U, Arendt T. The expression of cell cycle proteins in neurons and its relevance for Alzheimer's disease. Curr Drug Targets CNS Neurol Disord 2005;4:293-306. https://doi.org/10.2174/1568007054038175
- Lee HG, Casadesus G, Zhu X, et al. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem Int 2009;54:84-88. https://doi.org/10.1016/j.neuint.2008.10.013
- Moh C, Kubiak JZ, Bajic VP, et al. Cell cycle deregulation in the neurons of Alzheimer's disease. Results Probl Cell Differ 2011;53:565-576. https://doi.org/10.1007/978-3-642-19065-0_23
- Koseoglu MM, Norambuena A, Sharlow ER, et al. Aberrant neuronal cell cycle re-entry: The pathological confluence of Alzheimer's disease and brain insulin resistance, and its relation to cancer. J Alzheimers Dis 2019;67:1-11. https://doi.org/10.3233/JAD-180874
- McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses 1999;52:525-527. https://doi.org/10.1054/mehy.1997.0680
- Nagy Z. Cell cycle regulatory failure in neurones: causes and consequences. Neurobiol Aging 2000;21:761-769. https://doi.org/10.1016/s0197-4580(00)00223-2
- Arendt T. Alzheimer's disease as a disorder of mechanisms underlying structural brain self-organization. Neuroscience 2001;102:723-765. https://doi.org/10.1016/s0306-4522(00)00516-9
- Lopes JP, Oliveira CR, Agostinho P. Cell cycle re-entry in Alzheimer's disease: a major neuropathological characteristic? Curr Alzheimer Res 2009;6:205-212. https://doi.org/10.2174/156720509788486590
- Bonda DJ, Lee HP, Kudo W, et al. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev Mol Med 2010;12:e19. https://doi.org/10.1017/S146239941000150X
- Barrio-Alonso E, Hernández-Vivanco A, Walton CC, et al. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep 2018;8:14316. https://doi.org/10.1038/s41598-018-32708-4
- Olabiyi BF, Fleitas C, Zammou B et al. proNGF involvement in the adult neurogenesis dysfunction in Alzheimer's disease. Int J Mol Sci 2021;22:10744. https://doi.org/10.3390/ijms221910744
- Terreros-Roncal J, Moreno-Jiménez EP, Flor-García M, et al. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science 2021;374:1106-1113. https://doi.org/10.1126/science.abl516
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001; 12: 105-112. https://doi.org/10.1097/00041433-200104000-00003
- Naudí A, Cabré R, Jové M, et al. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int Rev Neurobiol 2015;122:133-189. https://doi.org/10.1016/bs.irn.2015.05.008
- Farooqui AA, Horrocks LA, Farooqui T. Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem Phys Lipids 2000;106:1-29. https://doi.org/10.1016/s0009-3084(00)00128-6
- Farooqui AA. Lipid mediators in the neural cell nucleus: Their metabolism, signaling, and association with neurological disorders. Neuroscientist 2009;15:392-407. https://doi.org/10.1177/1073858409337035
- Piomelli D. The challenge of brain lipidomics. Prostaglandins Lipid Mediat 2005;77: 23-34. https://doi.org/10.1016/j.prostaglandins.2004.09.006
- Piomelli D, Astarita G, Rapaka R. A neuroscientist's guide to lipidomics. Nature Rev Neurosci 2007; 8:743-754. https://doi.org/10.1038/nrn2233
- Pamplona R, Barja G, Portero-Otín M. Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: A homeoviscous-longevity adaptation? Ann N Y Acad Sci 2002;959:475-490. https://doi.org/10.1111/j.1749-6632.2002.tb02118.x
- Hulbert AJ, Pamplona R, Buffenstein R, et al. Life and death: Metabolic rate, membrane composition, and life span of animals. Physiol Rev 2007; 87:1175-1213. https://doi.org/10.1152/physrev.00047.2006
- Pamplona R, Ilieva E, Ayala V, Bellmunt MJ, et al. Maillard reaction versus other nonenzymatic modifications in neurodegenerative processes. Ann NY Acad Sci 2008;1126:315-319. https://doi.org/10.1196/annals.1433.014
- Moller M, Botti H, Batthyany C, et al. Direct measurement of nitric oxide and oxygen partitioning into liposomes and low density lipoprotein. J Biol Chem 2005;280:8850-8854. https://doi.org/10.1074/jbc.M413699200
- Gamliel A, Afri M, Frimer AA. Determining radical penetration of lipid bilayers with new lipophilic spin traps. Free Rad Biol Med 2008;44:1394-1405. https://doi.org/10.1016/j.freeradbiomed.2007.12.028
- Bielski BH, Arudi RL, Sutherland MW. A study of the reactivity of HO2/O2- with unsaturated fatty acids. J Biol Chem 1983;258:4759-4761. PMID: 6833274.
- Söderberg M, Edlund C, Kristensson K, et al. Lipid compositions of different regions of the human brain during aging. J Neurochem 1990;54:415-423. https://doi.org/10.1111/j.1471-4159.1990.tb01889.x
- Svennerholm L, Boström K, Helander CG, et al. Membrane lipids in the aging human brain. J Neurochem 1991;56:2051-2059. https://doi.org/10.1111/j.1471-4159.1991.tb03466.x
- Svennerholm L. Distribution and fatty acid composition of phosphoglycerides in normal human brain. J Lipid Res 1968;9:570-579. PMID: 4302302.
- Horrocks LA, VanRollins M, Yates AJ. Lipid changes in the ageing brain. In: The molecular basis of neuropathology. Davinson AN, Thompson RHS (Eds), Edward Arnold, 1981
- de Diego I, Peleg S, Fuchs B. The role of lipids in aging-related metabolic changes. Chem Phys Lipids 2019;222:59-69. https://doi.org/10.1016/j.chemphyslip.2019.05.005
- Johnson AA, Stolzing A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 2019;516:e13048. https://doi.org/10.1111/acel.13048
- McNamaraRK, Liu Y, Jandacek R, Rider T, et al. The aging human orbitofrontal cortex: Decreasing polyunsaturated fatty acid composition and associated increases in lipogenic gene expression and stearoyl-CoA desaturase activity. Prostaglandins, Leukotrienes, Essent Fatty Acids 2008;78:293-304. https://doi.org/10.1016/j.plefa.2008.04.00
- Keller JN, Dimayuga E, Chen Q, et al. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 2004;36: 2376-2391. https://doi.org/10.1016/j.biocel.2004.05.003
- Double KL, Dedov VN, Fedorow H, et al. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol Life Sci 2008;65:1669-1682.
- Porta EA. Advances in age pigment research. Arch Gerontol Geriat 1991;12:303-320. https://doi.org/10.1016/0167-4943(91)90036-p
- Jolly RD, Douglas BV, Davey PM, et al. Lipofuscin in bovine muscle and brain: A model for studying age pigment. Gerontology 1965;41:S283-S295. https://doi.org/10.1159/000213750
- Ottis P, Koppe K, Onisko B, et al. Human and rat brain lipofuscin proteome. Proteomics 2012;12:2445-2454. https://doi.org/10.1002/pmic.201100668
- Pamplona R, Dalfó E, Ayala V, et al. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation: effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem 2005; 280:21522-21530. https://doi.org/10.1074/jbc.M502255200
- Haughey NJ, Bandaru VV, Bae M, et al. Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochim Biophys Acta 2010; 1801: 878-886. https://doi.org/10.1016/j.bbalip.2010.05.003
- Frisardi V, Panza F, Seripa D, et al. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer's disease pathology. Progr Lipid Res 2011;50:313-330. https://doi.org/10.1016/j.plipres.2011.06.001
- Wood PL. Lipidomics of Alzheimer's disease: Current status. Alzheimer Res Ther 2012; 4: 5. https://doi.org/10.1186/alzrt103
- Kosicek M, Hecimovic S. Phospholipids and Alzheimer's disease: Alterations, mechanisms and potential biomarkers. Int J Mol Sci 2013;14:1310-1322. https://doi.org/10.3390/ijms14011310
- Touboul D, Gaudin M. Lipidomics of Alzheimer's disease. Bioanalysis 2014; 6: 541-561. https://doi.org/10.4155/bio.13.346
- Senanayake V, Goodenowe DB. Plasmalogen deficiency and neuropathology in Alzheimer’s disease: causation or coincidence? Alzheimers Dement 2019;5:524-532. https://doi.org/10.1016/j.trci.2019.08.003
- Brosche T, Platt D. The biological significance of plasmalogens in defense against oxidative damage. Exp Gerontol 1998;33:363-369. https://doi.org/10.1016/s0531-5565(98)00014-x
- Martínez A, Portero-Otin M, Pamplona R, et al. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 2010; 20:281-297. https://doi.org/10.1111/j.1750-3639.2009.00326.x
- Sultana R, Perluigi M, Butterfield AD. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Rad Biol Med 2013;62:157-169. https://doi.org/10.1016/j.freeradbiomed.2012.09.027
- Benseny-Cases N, Klementieva O, Cotte M, et al. Microspectroscopy (μFTIR) reveals co-localization of lipid oxidation and amyloid plaques in human Alzheimer disease brains. Anal Chem 2014;86:12047-12054. https://doi.org/10.1021/ac502667b
- Evans RM, Hui S, Perkins A, et al. Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology 2004;62: 1869-1871. https://doi.org/10.1212/01.wnl.0000125323.15458.3f
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 2009;10:333-344. https://doi.org/10.1038/nrn2620
- Hope C, Mettenburg J, Gonias SL, et al. Functional analysis of plasma α2-macroglobulin from Alzheimer’s disease patients with the A2M intronic deletion. Neurobiol Dis 2003;14:504-512. https://doi.org/10.1016/j.nbd.2003.08.005
- Zappia M, Manna I, Serra P, et al. Increased risk for Alzheimer disease with the interaction of MPO and A2M polymorphisms. Arch Neurol 2004;61:341-344. https://doi.org/10.1001/archneur.61.3.341
- Marzolo MP, Bu G. Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin Cell Dev Biol 2009;20:191-200. https://doi.org/10.1016/j.semcdb.2008.10.005
- Jones EM, Dubey M, Camp PJ, et al. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemistry 2012;51:2539-2550. https://doi.org/10.1021/bi201857v
- Morgan K. The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol 2011;37:353-357. https://doi.org/10.1111/j.1365-2990.2011.01181.x
- D Bruce K, Tang M, Reigan P, et al. Genetic variants of lipoprotein lipase and regulatory factors associated with Alzheimer's disease risk. Int J Mol Sci 2020;21:8338 https://doi.org/10.3390/ijms21218338
- Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018;141:2181-2193. https://doi.org/10.1093/brain/awy146
- Robinson AC, Davidson YS, Roncaroli F, et al. Influence of APOE genotype in primary age-related tauopathy. Acta Neuropathol Commun 2020; 8:215. https://doi.org/10.1186/s40478-020-01095-1
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science 2010;327:46-50. https://doi.org/10.1126/science.1174621
- Sezgin E, Levental I, Mayor S, et al. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 2017;18:361-374. https://doi.org/10.1038/nrm.2017.16
- Tatulian SA, Qin S, Pande AH, et al. Positioning membrane proteins by novel protein engineering and biophysical approaches. J Mol Biol 2005;351:939-947. https://doi.org/10.1016/j.jmb.2005.06.080
- Tun H, Marlow L, Pinnix I, et al. Lipid rafts play an important role in Aβ biogenesis by regulating the β-secretase pathway. J Mol Neurosci 2002;19:31-35. https://doi.org/10.1007/s12031-002-0007-5
- Ehehalt R, Keller P, Haass C, et al. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 2003;160:113-123. https://doi.org/10.1083/jcb.200207113
- Kawarabayashi T, Shoji M, Younkin LH, et al. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apoilipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 2004;24:3801-3809. https://doi.org/10.1523/JNEUROSCI.5543-03.2004
- Williamson R, Usardi A, Hanger DP, et al. Membrane-bound β-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J 2008;22:1552-1559. https://doi.org/10.1096/fj.07-9766com
- Williamson R, Sutherland C. Neuronal membranes are key to the pathogenesis of Alzheimer's disease: the role of both raft and non-raft membrane domains. Curr Alzheimer Res 20118:213-221. https://doi.org/10.2174/156720511795256008
- Fabiani C, Antollini SS. Alzheimer's disease as a membrane disorder: spatial cross-talk among β-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front Cell Neurosci 2019;13:309. https://doi.org/10.3389/fncel.2019.00309
- Fabelo N, Martín V, Marín R, et al. Evidence for premature lipid raft aging in APP/PS1 double-transgenic mice, a model of familial Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:868-881. https://doi.org/10.1097/NEN.0b013e31826be03c
- Takahashi RH, Tobiume M, Sato Y, et al. Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer's disease brain. Neuropathology 2011;31:208-214. https://doi.org/10.1111/j.1440-1789.2010.01158.x
- Takahashi RH, Yokotsuka M, Tobiume M, et al. Accumulation of cellular prion protein within β-amyloid oligomer plaques in aged human brains. Brain Pathol 2021;31:e12941. https://doi.org/10.1111/bpa.12941
- Díaz M, Fabelo N, Ferrer I, et al. “Lipid raft aging” in the human frontal cortex during nonpathological aging: gender influences and potential implications in Alzheimer's disease. Neurobiol Aging 2018;67:42-52. https://doi.org/10.1016/j.neurobiolaging.2018.02.022
- Fabelo N, Martín V, Marín R, et al. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer's disease and facilitates APP/BACE1 interactions. Neurobiol Aging 2014;35:1801-1812. https://doi.org/10.1016/j.neurobiolaging.2014.02.005
- Díaz M, Fabelo N, Martín V, et al. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer's disease. J Alzheimers Dis 2015;43:1185-1198. https://doi.org/10.3233/JAD-141146
- Chi EY, Ege C, Winans A, et al. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer's disease amyloid-beta peptide. Proteins 2008;72:1-24. https://doi.org/10.1002/prot.21887
- Vetrivel KS, Thinarkaran G. Membrane rafts in Alzheimer's disease beta-amyloid production. Biochem Biophys Acta - Molecular and Cell Biology of Lipids 2010;1801:860-867. https://doi.org/10.1016/j.bbalip.2010.03.007
- Relini A, Marano N, Gliozzi A. Probing the interplay between amyloidogenic proteins and membranes using lipid monolayers and bilayers. Adv Colloid Interface Sci 2014;207:81-92. https://doi.org/10.1016/j.cis.2013.10.015
- Georgieva ER, Xiao S, Borbat PP, et al. Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats. Biophys J 2014;107:1441-1452. https://doi.org/10.1016/j.bpj.2014.07.046
- Majewski J, Jones EM, Vander Zanden CM, et al. Lipid membrane templated misfolding and self-assembly of intrinsically disordered tau protein. Sci Rep 2020;10:13324. https://doi.org/10.1038/s41598-020-70208-6
- Künze G, Barré P, Scheidt HA, et al. Binding of the three-repeat domain of tau to phospholipid membranes induces an aggregated-like state of the protein. Biochim Biophys Acta Biomembr 2012; 1818: 2302–2313. https://doi.org/10.1016/j.bbamem.2012.03.019
- Elbaum-Garfinkle S, Ramlall T, Rhoades E. The role of the lipid bilayer in tau aggregation. Biophys J 2010; 98: 2722-2730. https://doi.org/10.1016/j.bpj.2010.03.013
- Chad A Sallaberry CA, Voss BJ, et al. Tau and membranes: interactions that promote folding and condensation. Front Cell Dev Biol 2021;9:725241. https://doi.org/10.3389/fcell.2021.725241
- Ferrer I, Andrés-Benito P, Ausín K, et al. Dysregulated protein phosphorylation: a determining condition in the continuum of brain aging and Alzheimer’s disease. Brain Pathol 2021; 31:e12996. https://doi.org/10.1111/bpa.12996
- Quest AF, Leyton L, Párraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol 2004;82:129-144. https://doi.org/10.1139/o03-071
- van Helmond ZK, Scott Miners J, Bednall E, et al. Caveolin-1 and -2 and their relationship to cerebral amyloid angiopathy in Alzheimer's disease. Neuropathol Appl Neurobiol 2007;33:317-327. https://doi.org/10.1111/j.1365-2990.2006.00815.x
- Ramírez CM, González M, Díaz M, et al. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer's disease. Mol Cell Neurosci 2009;42:172-183. https://doi.org/10.1016/j.mcn.2009.07.001
- Puangmalai N, Bhatt N, Montalbano M, et al. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer's disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis 2020;11:314. https://doi.org/10.1038/s41419-020-2503-3
- de Groot NS, Burgas MT. Is membrane homeostasis the missing link between inflammation and neurodegenerative diseases? Cell Mol Life Sci 2015;72:4795-805. https://doi.org/10.1007/s00018-015-2038-4
- Krigman MR, Feldman RG, Bensch K. Alzheimer’s presenile dementia. A histochemical and electron microscopic study. Lab Invest 1965;14:381-396. PMID: 14281440.
- Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 2001;21:3017-3023. https://doi.org/10.1523/JNEUROSCI.21-09-03017.2001
- Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. J Alzheimer’s Dis 2006; 9:119-126. https://doi.org/10.3233/jad-2006-9204
- Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis 2010; 20: 20:S569-578. https://doi.org/10.3233/JAD-2010-100357
- Cheng Y, Bai F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci 2018;12:163. https://doi.org/10.3389/fnins.2018.00163
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63:8-20. https://doi.org/10.1016/j.mehy.2003.12.045
- Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 2018; 62:1403-1416. https://doi.org/10.3233/JAD-170585
- Hauptmann S, Keil U, Scherping I, et al. Mitochondrial dysfunction in sporadic and genetic Alzheimer's disease. Exp Gerontol 2006;41:668-673. https://doi.org/10.1016/j.exger.2006.03.012
- Grimm A, Friedland K, Eckert A. Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer's disease. Biogerontology 2016;17:281-296. https://doi.org/10.1007/s10522-015-9618-4
- Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 2020;15:30. https://doi.org/10.1186/s13024-020-00376-6
- Banks B, Ingram TL, Chakrabarti L. ATP synthase and Alzheimer's disease: putting a spin on the mitochondrial hypothesis Aging (Albany NY) 2020;12:16647-16662. https://doi.org/10.18632/aging.103867
- Terada T, Therriault J, Kang MSP, et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer's disease. Mol Neurodegener 2021;16:28. https://doi.org/10.1186/s13024-021-00448-1
- Kawamata H, Manfredi G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J Cell Biol 2017;216:3917-3929. https://doi.org/10.1083/jcb.201709172
- Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta - Mol Basis Dis 2014; 1842:1219-1231. https://doi.org/10.1016/j.bbadis.2013.09.010
- Hoekstra JG, Hipp MJ, Montine TJ, et al. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol 2016;80:301-306. https://doi.org/10.1002/ana.24709
- Terni B, Boada J, Portero-Otin M, Pamplona R, et al. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer's disease pathology. Brain Pathol 2010;20:222-233. https://doi.org/10.1111/j.1750-3639.2009.00266.x
- Busciglio J, Pelsman A, Wong C, et al. Altered metabolism of the amyloid precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002;33: 677-688. https://doi.org/10.1016/s0896-6273(02)00604-9
- Kandimalla R, Manczak M, Yin X, et al. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 2018;27:30-40. https://doi.org/10.1093/hmg/ddx381
- Eckert A, Schulz KL, Rhein V, et al. Convergence of amyloid-beta and tau on mitochondria in vivo. Mol Neurobiol 2010;41:107-114. https://doi.org/10.1007/s12035-010-8109-5
- Wilkins HM, Swerdlow RH. Mitochondrial links between brain aging and Alzheimer's disease. Transl Neurodegener 2021;10:33. https://doi.org/10.1186/s40035-021-00261-2
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol 2000;19:81-88. https://doi.org/10.1186/s40035-021-00261-2
- Eysert F, Kinoshita PF, Mary A, et al. Molecular dysfunctions of mitochondria-associated membranes (MAMs) in Alzheimer's disease. Int J Mol Sci 2020;21:9521. https://doi.org/10.3390/ijms21249521
- Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 2013;55:26-36. https://doi.org/10.1016/j.mcn.2012.07.011
- Area-Gomez E, Schon E A. Mitochondria-associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 2016;38:90-96. https://doi.org/10.1016/j.gde.2016.04.006
- Area-Gomez E, Schon EA. On the pathogenesis of Alzheimer’s disease: the MAM hypothesis. FASEB J 2017;31:864-867. https://doi.org/10.1096/fj.201601309
- Pamplona R. Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta 2008; 1777:1249-1262. https://doi.org/10.1016/j.bbabio.2008.07.003
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2013; 2013:316523. https://doi.org/10.1155/2013/316523
- Thomas DD, Ridnour L, Donzelli S, et al. The chemistry of protein modifications elicited by nitric oxide and related nitrogen oxides. In: Redox proteomics: from protein modifications to cellular dysfunction and diseases. Dalle-Done I, Scaloni A, Butterfield DA (eds). JohnWiley & Sons, 2006: 25-58.
- Cabré R, Naudí A, Dominguez-Gonzalez M, et al. Sixty years old is the breakpoint of human frontal cortex aging. Free Radic Biol Med 2017;103:14-22. https://doi.org/10.1016/j.freeradbiomed.2016.12.010
- Domínguez M, de Oliveira E, Odena MA, et al. Redox proteomic profiling of neuroketal-adducted proteins in human brain: Regional vulnerability at middle age increases in the elderly. Free Radic Biol Med 2016;95:1-15. https://doi.org/10.1016/j.freeradbiomed.2016.02.034
- Choi WT, Tosun M, Jeong HH, et al. Metabolomics of mammalian brain reveals regional differences. BMC Syst Biol 2018;12:127. https://doi.org/10.1186/s12918-018-0644-0
- Cabré R, Jové M, Naudí A, et al. Specific metabolomics adaptations define a differential regional vulnerability in the adult human cerebral cortex. Front Mol Neurosci 2016;9:138. https://doi.org/10.3389/fnmol.2016.00138
- Domínguez-González M, Puigpinós M, Jové M, et al. Regional vulnerability to lipoxidative damage and inflammation in normal human brain aging. Exp Gerontol 2018;111:218-228. https://doi.org/10.1016/j.exger.2018.07.023
- Srikanth V, Maczurek A, Phan T, et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol Aging 2011;32:763-777. https://doi.org/10.1016/j.neurobiolaging.2009.04.016
- Chambers A, Bury JJ, Minett T, et al. Advanced glycation end product formation in human cerebral cortex increases with Alzheimer-type neuropathologic changes but is not independently associated with dementia in a population-derived aging brain cohort. J Neuropathol Exp Neurol 2020;79:950-958. https://doi.org/10.1093/jnen/nlaa064
- Korolainen MA, Auriola S, Nyman TA, et al. Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol Dis 2005;20:858-870. https://doi.org/10.1016/j.nbd.2005.05.021
- Korolainen MA, Goldsteins G, Nyman TA, et al. Oxidative modification of proteins in the frontal cortex of Alzheimer’s disease brain. Neurobiol Aging 2006;27:42-53. https://doi.org/10.1016/j.neurobiolaging.2004.11.010
- Butterfield DA, Sultana R. Redox proteomics: understanding oxidative stress in the progression of age-related neurodegenerative disorders. Expert Rev Proteomics 2008; 5:157-160. https://doi.org/10.1586/14789450.5.2.157
- Butterfield DA, Abdul HM, Newman S, et al. Redox proteomics in some age-related neurodegenerative disorders or models thereof. NeuroRx 2006; 3:344-357. https://doi.org/10.1016/j.nurx.2006.05.003
- Butterfield DA, Gnjec A, Poon HF, et al. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: an initial assessment. J Alzheimers Dis 2006;10:391-397. https://doi.org/10.3233/jad-2006-10407
- Butterfield DA, Poon HF, St Clair D, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis 2006;22:223-232. https://doi.org/10.1016/j.nbd.2005.11.002
- Sultana R, Boyd-Kimball D, Poon HF, et al. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 2006;27:1564-1576. https://doi.org/10.1016/j.neurobiolaging.2005.09.021
- Sultana R, Poon HF, Cai J, et al. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis 2006;22:76-87. https://doi.org/10.1016/j.nbd.2005.10.004
- Reed T, Perluigi M, Sultana R, Pierce WM, et al. Proteomic identification of 4-hydroxy-2-nonenal modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis 2008;30:107-120. https://doi.org/10.1016/j.nbd.2007.12.007.
- Reed TT, Pierce WM Jr, Turner DM, et al. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J Cell Mol Med 2009;13:2019-2029. https://doi.org/10.1111/j.1582-4934.2008.00478.x
- Cabiscol E, Ros J. Oxidative damage to proteins: structural modifications and consequences in cell function. In: Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases. I Dalle-Donne, A Scaloni, DA Butterfield (eds), John Wiley & Sons: Hoboken, NJ. 2006; pp. 399-471.
- Zabel M, Nackenoff A, Kirsch WM, et al. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer's disease brain: A meta-analysis in human pathological specimens. Free Radic Biol Med 2018;115:351-360. https://doi.org/10.1016/j.freeradbiomed.2017.12.016
- Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001;60:759-767. https://doi.org/10.1093/jnen/60.8.759
- Nunomura A, Castellani RJ, Zhu X, et al. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 2006;65:631-641. https://doi.org/10.1097/01.jnen.0000228136.58062.bf
- Nunomura A, Perry G, Pappolla MA, et al. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol 2000;59:1011-1017. https://doi.org/10.1093/jnen/59.11.1011
- Nunomura A, Chiba S, Lippa CF, et al. Neuronal RNA oxidation is a prominent feature of familial Alzheimer's disease. Neurobiol Dis 2004;17:108-113. https://doi.org/10.1016/j.nbd.2004.06.003
- Smith MA, Sayre LM, Monnier VM, et al. Oxidative posttranslational modifications in Alzheimer disease. A possible pathogenic role in the formation of senile plaques and neurofibrillary tangles. Mol Chem Neuropathol 1996;28:41-48. https://doi.org/10.1007/BF02815203
- Nunomura A, Honda K, Takeda A, et al. Oxidative damage to RNA in neurodegenerative diseases. J Biomed Biotechnol 2006;2006: 82323. https://doi.org/10.1155/JBB/2006/82323
- Tamagno E, Guglielmotto M, Vasciaveo V, et al. Oxidative stress and beta- amyloid in Alzheimer's disease. Which comes first: the chicken or the egg? Antioxidants 2021;10:1479. https://doi.org/10.3390/antiox10091479
- Wang X, Wang W, Li L, et al. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 2014; 1842:1240-1247. https://doi.org/10.1016/j.bbadis.2013.10.015
- Jové M, Pradas I, Dominguez-Gonzalez M, et al. Lipids and lipoxidation in human brain aging. Mitochondrial ATP-synthase as a key lipoxidation target. Redox Biol 2019;23:101082. https://doi.org/10.1016/j.redox.2018.101082
- Jové M, Mota-Martorell N, Torres P, et al. The causal role of lipoxidative damage in mitochondrial bioenergetic dysfunction linked to Alzheimer's disease pathology. Life 2021;11:388. https://doi.org/10.3390/life11050388
- Ferrer I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer's disease. J Bioenerg Biomembr 2009;41:425-431. https://doi.org/10.1007/s10863-009-9243-5
- Münch G, Gasic-Milenkovic J, Arendt T. Effect of advanced glycation endproducts on cell cycle and their relevance for Alzheimer's disease. J Neural Transm Suppl 2003;65:63-71. https://doi.org/10.1007/978-3-7091-0643-3_4
- Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol 2008;3:41-66. https://www.doi.org/10.1146/annurev.pathmechdis.2.010506.092044
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 2009;64:110-122. https://doi.org/10.1016/j.neuron.2009.08.039
- Jenkinson ML, Bliss MR, Brain AT, Scott DL. Rheumatoid arthritis and senile dementia of the Alzheimer’s type. Br J Rheumatol 1989;28:86-88. https://doi.org/10.1093/rheumatology/28.1.86-b
- McGeer PL, McGeer E, Rogers J, Sibley J. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990;335:1037. https://doi.org/10.1016/0140-6736(90)91101-f
- Myllykangas-Luosujarvi R, Isomaki H. Alzheimer’s disease and rheumatoid arthritis. Br J Rheumatol 1994;33:501-502. https://doi.org/10.1093/rheumatology/33.5.501
- Breitner JC, Welsh KA, Helms MJ, et al. Delayed onset of Alzheimer’s disease with non-steroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging 1995;16:523-530. https://doi.org/10.1016/0197-4580(95)00049-k
- Stewart WF, Kawas C, Corrada M, et al. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997;48:626-632. https://doi.org/10.1212/wnl.48.3.626
- Mackenzie IRA, Muñoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 1998;50:986-990. https://doi.org/10.1212/wnl.50.4.986
- Zandi PP, Anthony JC, Hayden KM, et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: The Cache County Study. Neurology 2002;59:880-886. https://doi.org/10.1212/wnl.59.6.880
- Vlad SC, Miller DR, Kowall NW, et al. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008;70:1672-1677. 10.1212/01.wnl.0000311269.57716.63.
- Terzi M, Altun G, Şen S, et al. The use of non-steroidal anti-inflammatory drugs in neurological diseases. J Chem Neuroanat 2018;87:12-24. https://doi.org/10.1016/j.jchemneu.2017.03.003
- Kaduševičius E. Novel applications of NSAIDs: insight and future perspectives in cardiovascular, neurodegenerative, diabetes and cancer disease therapy. Int J Mol Sci 2021;22:6637. https://doi.org/10.3390/ijms22126637
- Wilberding A, Morimoto K, Satoh H, et al. Multiple cytokines are involved in the early events leading to the Alzheimer's disease pathology.Tottori Rinsho Kagaku Kenkyukai Shi 2008;1:359-373. PMID: 22586434.
- Sajdel-Sulkowska EM, Marotta CA. Alzheimer's disease brain: alterations in RNA levels and in a ribonuclease-inhibitor complex. Science 1984;225: 947-949. https://doi.org/10.1126/science.6206567
- Langstrom NS, Anderson JP, Lindroos HG, et al. Alzheimer's disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res 1989;5:259-269. https://doi.org/10.1016/0169-328x(89)90060-0
- da Silva AM, Payão SL, Borsatto B, et al. Quantitative evaluation of rRNA in Alzheimer’s disease. Mech Ageing Dev 2000;120: 57-64. https://doi.org/10.1016/s0047-6374(00)00180-9
- Ferrer I. Differential expression of phosphorylated translation initiation factor 2 alpha in Alzheimer's disease and Creutzfeldt-Jakob's disease. Neuropathol Appl Neurobiol 2002;28: 441-451. https://doi.org/10.1046/j.1365-2990.2002.t01-1-00410.x
- Shan X, Tashiro H, Lin CL. The identification and characterization of oxidized RNAs in Alzheimer's disease. J Neurosci 2003;23: 4913-4921. https://doi.org/10.1523/JNEUROSCI.23-12-04913.2003
- Li X, An WL, Alafuzoff I, et al. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport 2004;15: 2237-2240. https://doi.org/10.1097/00001756-200410050-00019
- Nunomura A, Perry G. RNA and Oxidative Stress in Alzheimer's Disease: Focus on microRNAs. Oxid Med Cell Longev 2020;2020:2638130. https://doi.org/10.1155/2020/2638130
- Dönmez-Altuntas H, Akalain H, Karaman Y, et al. Evaluation of the nucleolar organizer regions in Alzheimer’s disease. Gerontology 2005;51: 297-301. https://doi.org/10.1159/000086365
- Honda K, Smith MA, Zhu X, et al. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron.J Biol Chem 2005;280:20978-20986. https://doi.org/10.1074/jbc.M500526200
- Ding Q, Markesbery WR, Chen Q, et al. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci 2005;25: 9171-9175. https://doi.org/10.1523/JNEUROSCI.3040-05.2005
- Ding Q, Markesbery WR, Cecarini V, et al. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res 2006;31:705-710. https://doi.org/10.1007/s11064-006-9071-5
- Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer's disease. Neurobiol Aging 2006 27: 657-662. https://doi.org/10.1016/j.neurobiolaging.2005.03.022
- Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer disease. Neuron 2018;99:925-940.e7. https://doi.org/10.1016/j.neuron.2018.07.039
- Hernández-Ortega K, Garcia-Esparcia P, Gil L, et al. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol 2016;26: 593-605. https://doi.org/10.1111/bpa.12335
- Di Domenico F, Sultana R, Barone E, et al. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer's disease subjects. J Proteomics 2011;74:1091-1103. https://doi.org/10.1016/j.jprot.2011.03.033
- Xia Q, Cheng D, Duong DM, et al. Phosphoproteomic analysis of human brain by calcium phosphate precipitation and mass spectrometry. J Proteome Res 2008;7:2845-2851. https://doi.org/10.1021/pr8000496
- Tan H, Wu Z, Wang H, et al. Refined phosphopeptide enrichment by phosphate additive and the analysis of human brain phosphoproteome. Proteomics 2015;15:500-507. https://doi.org/10.1002/pmic.201400171
- Triplett JC, Swomley AM, Cai J, et al. Quantitative phosphoproteomic analyses of the inferior parietal lobule from three different pathological stages of Alzheimer's disease. J Alzheimers Dis 2016;49:45-62. https://doi.org/10.3233/JAD-150417
- Dammer EB, Lee AK, Duong DM, et al. Quantitative phosphoproteomics of Alzheimer's disease reveals cross-talk between kinases and small heat shock proteins. Proteomics 2015;15:508-519. https://doi.org/10.1002/pmic.201400189
- Sathe G, Mangalaparthi KK, Jain A, et al. Multiplexed phosphoproteomic study of the brain in patients with Alzheimer's disease and age-matched cognitively healthy controls. OMICS 2020;24:2016-2227. https://doi.org/10.1089/omi.2019.0191
- Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer's disease progression. Neuron 2020;105: 975-991.e7. https://doi.org/10.1016/j.neuron.2019.12.015
- Mérida I, Avila-Flores A, Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem J 2008;409:1-18. https://doi.org/10.1042/BJ20071040
- Almena M, Mérida I. Shaping up the membrane: diacylglycerol coordinates spatial orientation of signalling. Trends Biochem Sci 2011;36:593-603. https://doi.org/10.1016/j.tibs.2011.06.005
- Sakane F, Hoshino F, Murakami C. New era of diacylglycerol kinase, phosphatidic acid and phosphatidic acid-binding protein. Int J Mol Sci 2020;21:6794. https://doi.org/10.3390/ijms21186794
- Ekinci FJ, Shea TB. Free PKC catalytic subunits (PKM) phosphorylate tau via a pathway distinct from that utilized by intact PKC. Brain Res 1999;850:207-216. https://doi.org/10.1016/s0006-8993(99)02146-0
- Chan RB, Oliveira TG, Cortes EP, et al. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem 2012;287:2678-2688. https://doi.org/10.1074/jbc.M111.274142
- Wood PL, Medicherla S, Sheikh N, et al. Targeted lipidomics of frontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer's disease: validation of DAG accumulation early in the pathophysiology of Alzheimer's disease. J Alzheimers Dis 2015;48:537-546. https://doi.org/10.3233/JAD-150336
- Pinheiro L, Faustino C. Therapeutic strategies targeting amyloid-β in Alzheimer's disease. Curr Alzheimer Res 2019;16:418-452. https://doi.org/10.2174/1567205016666190321163438
- icoll JA, Wilkinson D, Holmes C, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003;9: 448-452. https://doi.org/10.1038/nm840
- Nicoll JAR, Buckland GR, Harrison CH, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer's disease. Brain 2019; 142: 2113-2126. https://doi.org/10.1093/brain/awz142
- Loureiro JC, Pais MV, Stella F, et al. Passive antiamyloid immunotherapy for Alzheimer’s disease. Curr Opin Psychiatry 2020; 33: 284-291. https://doi.org/10.1097/YCO.0000000000000587
- Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 2018; 378, 321-330. https://doi.org/10.1056/NEJMoa1705971
- van Bokhoven P, de Wilde A, Vermunt L, et al. The Alzheimer’s disease drug development landscape. Alzheimers Res Ther 2021;13:186. https://doi.org/10.1186/s13195-021-00927-z
- Abushouk AI, Elmaraezy A, Aglan A, et al. Bapineuzumab for mild to moderate Alzheimer’s disease: A meta-analysis of randomized controlled trials. BMC Neurol 2017;17:66. https://doi.org/10.1186/s12883-017-0850-1
- Ratti E, Kong J, O’Gorman J, Rajagovindan R, et al. Baseline characteristics from TANGO: Phase 2 study to evaluate gosuranemab (BIIB092) in patients with early Alzheimer’s disease. Alzheimers Dement 2020;16, e044910.
- Walsh S, Merrick R, Milne R, et al. Aducanumab for Alzheimer’s disease? BMJ 2021;374: n1682. https://doi.org/10.1136/bmj.n1682
- Querol-Vilaseca M, Sirisi MS, Molina-Porcel L, et al. Neuropathology of a patient with Alzheimer disease treated with low doses of Verubecestat. Neuropathol Appl Neurobiol 2022;48:e12781. https://doi.org/10.1111/nan.12781
- Han P, Shi J.A. Theoretical analysis of the synergy of amyloid and tau in Alzheimer's disease. J Alzheimers Dis 2016;52:1461-70. https://doi.org/10.3233/JAD-151206
- Sherman MA, LaCroix M, Amar F, et al. Soluble conformers of Aβ and Tau alter selective proteins governing axonal transport. J Neurosci 2016;36:9647-9658. https://doi.org/10.1523/JNEUROSCI.1899-16.2016
- Kim CM, Montal V, Diez I, et al. Network interdigitations of Tau and amyloid-beta deposits define cognitive levels in aging. Hum Brain Mapp 2021;42:2990-3004. https://doi.org/10.1002/hbm.25350
- Braczynski AK, Schulz JB, Bach JP. Vaccination strategies in tauopathies and synucleinopathies. J Neurochem 2017;143:467-488. https://doi.org/10.1111/jnc.14207
- Congdon EE, Sigurdson EM. Tau-targeting therapies for Alzheimer’s disease. Nature Rev 2018;14: 399-415. https://doi.org/10.1038/s41582-018-0013-z
- Medina M. An overview on the clinical development of tau-based therapies. Int J Mol Sci 2018;19:1160. https://doi.org/10.3390/ijms19041160
- Jadhav S, Avila J, Schöll M, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun 2019;7:22. https://doi.org/10.1186/s40478-019-0664-z
- Mullard A. Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat Rev Drug Discov 2020;20:3-5. https://doi.org/10.1038/d41573-020-00217-7
- Loera-Valencia R, Piras A, Ismail MAM, et al.Targeting Alzheimer’s disease with gene and cell therapies. J Intern Med 2018; 284:2-36. https://doi.org/10.1111/joim.12759
- Dunbar CE, High KA, Joung JK, et al. Gene therapy comes of age. Science 2018; 359:eaan4672. https://doi.org/10.1126/science.aan4672
- Lennon MJ, Rigney G, Raymont V, et al. Genetic therapies for Alzheimer’s disease: a scoping review. J Alz Dis 2021; 84: 491-504. https://doi.org/10.3233/JAD-215145
- Tuszynski MH, Thal L, Pay M, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 2005; 11: 551-555. https://doi.org/10.1038/nm1239
- Eriksdotter-Jönhagen M, Linderoth B, Lind G, et al. Encapsulated cell biodelivery of nerve growth factor to the basal forebrain in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord 2012;33:18-28. https://doi.org/10.1159/000336051
- Rafii MS, Tuszynski MH, Thomas RG, et al. Adeno-associated viral vector (Serotype 2)-nerve growth factor for patients with Alzheimer disease: A randomized clinical trial. JAMA Neurol 2018; 75: 834-841. https://doi.org/10.1001/jamaneurol.2018.0233
- Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 2020; 27:18. https://doi.org/10.1186/s12929-019-0609-7
- Cummings J, Lee G, Zhong K, Fonseca J, et al. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement 2021;7:e12179. https://doi.org/10.1002/trc2.12179
- McDade E, Llibre-Guerra JJ, Holtzman DM, et al. The informed road map to prevention of Alzheimer Disease: A call to arms. Mol Neurodegener 2021;16:49. https://doi.org/10.1186/s13024-021-00467-y
- Cummings J. Disease modification is not all — we need symptomatic therapies for Alzheimer disease. Nature Rev Neurol 2022;18:3-4. https://doi.org/10.1038/s41582-021-00591-9
- Gotz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci 2018;9:583-598. https://doi.org/10.1038/s41583-018-0054-8
- Vitek MP, Araujo JA, Fossel M, et al. Translational animal models for Alzheimer's disease: An Alzheimer's Association Business Consortium Think Tank. Alzheimers Dement 2021;6:e12114. https://doi.org/10.1002/trc2.12114
- Mckean NE, Handley RR, Snell RG. A review of the current mammalian models of Alzheimer’s disease and challenges that need to be overcome. Int J Mol Sci 2021;22:13168. https://doi.org/10.3390/ijms222313168
- Arendt T. Alzheimer's disease as a disorder of dynamic brain self-organization. Prog Brain Res 2005;147:355-378. https://doi.org/10.1016/S0079-6123(04)47025-3
- Swerdlow RH. Alzheimer's disease pathologic cascades: who comes first, what drives what. Neurotox Res 2012;22:182-194. https://doi.org/10.1007/s12640-011-9272-9
- Sharpee TO, Destexhe A, Kawato M, et al. 25th Annual Computational Neuroscience meeting: CNS-2016. BMC Neurosci 2016;17 Suppl 1:54. https://doi.org/10.1186/s12868-016-0283-6
- Bashtrykov P, Jeltsch A. Epigenome editing in the brain. Adv Exp Med Biol 2017;978:409-424. https://doi.org/10.1007/978-3-319-53889-1_21
- Mancioppi G, Fiorini L, Timpano Sportiello M, et al. Novel technological solutions for assessment, treatment, and assistance in mild cognitive impairment. Front Neuroinform 2019;13:58. https://doi.org/10.3389/fninf.2019.00058
- Romanella SM, Roe D, Paciorek R, et al. Sleep, noninvasive brain stimulation, and the aging brain: challenges and opportunities. Ageing Res Rev 2020;61:101067. https://doi.org/10.1016/j.arr.2020.101067
- El-Atab N, Shaikh SF, Hussain MM. Nano-scale transistors for interfacing with brain: design criteria, progress and prospect. Nanotechnology 2019;30:442001. https://doi.org/10.1088/1361-6528/ab3534
- Koutentakis D, Pilozzi A, Huang X. Designing socially assistive robots for Alzheimer’s disease and related dementia patients and their caregivers: where we are and where we are headed. Healthcare 2020;8:73. https://doi.org/10.3390/healthcare8020073
- Ling TS, Chandrasegaran S, Xuan LZ, et al. The potential benefits of nanotechnology in treating Alzheimer's disease. Biomed Res Int 2021;2021:5550938. https://doi.org/10.1155/2021/5550938
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
|