|
|
|
Free Neuropathology 3:4 (2022) |
|
Review |
|
Neurooncology: 2022 update |
|
Pieter Wesseling1,2, Jacob S. Rozowsky2 |
|
1 Department of Pathology, Amsterdam University Medical Centers/VUmc, Brain Tumor Center Amsterdam, De Boelelaan 1117, 1081HV Amsterdam, The Netherlands |
|
Corresponding author: |
|
Submitted: 14 February 2022 Accepted: 23 February 2022 Copyedited by: Henry Robbert Published: 24 February 2022 |
|
Keywords: Brain tumor, Neuropathology, Molecular diagnostics, Glioblastoma, Medulloblastoma |
|
Abstract This ‘Neurooncology: 2022 update’ presents topics that were selected by the authors as top ten discoveries published in 2021 in the broader field of neurooncological pathology. This time, the spectrum of topics includes: papers with a direct impact on daily diagnostic practice of CNS tumors in general and with information on how to improve grading of meningiomas; studies shedding new light on the oncogenesis of gliomas (in particular ‘optic gliomas’ and H3-mutant gliomas); several ‘multi-omic’ investigations unraveling the intra-tumoral heterogeneity of especially glioblastomas further; a study indicating the potential of ‘repurposing’ Prozac® for the treatment of glioblastomas; liquid biopsy using CSF for assessment of residual medulloblastoma. In the last part of this review some other papers are mentioned that didn’t make it to this (quite subjective) top ten list. Introduction For the third year in a row, the first author of this paper was invited to contribute a review on the top ten discoveries in neurooncology published in the previous year. Rather than checking over 10,000 papers on tumors of the central nervous system (CNS) published in 2021 (as was done for the previous review), he now restricted himself to screening the higher-ranked neurooncology and neuropathology journals and the top-journals in oncology and science with a broader scope. Furthermore, he teamed up with the last author, a young Fulbright scholar from the USA that he supervises for a period of 9 months in the Netherlands. The last author mainly used a Twitter®-based approach for identifying promising candidates for our list of top ten discoveries in 2021. Interestingly, there was quite some overlap between the results of these different search strategies (but more research is needed for sorting out if Twitter® is a reliable source of information in this respect, also because it appears that in other areas of life the information shared by tweets can cause chaos rather than order). The final list of topics selected by the authors as very interesting and/or very important for the broader field of neurooncological pathology this time looks like this:
Of note, all the papers just mentioned became officially available in 2021, but the printed version may have lagged somewhat behind. Also, and like in previous years, one has to realize that there is of course quite a subjective component in the selection process that resulted in this list. For example, the strong interest of the last author in computational biology aspects of CNS tumors may well have played a role in the selection of topics 5 and 6 especially. To make up to some degree for this rather subjectively composed bouquet of papers, in the Discussion section of this review some papers/topics are mentioned that didn’t make it to this Top ten list but that certainly are interesting as well. Hopefully, this review thereby again helps to appreciate the wealth of (sometimes mind-boggling) information provided by the papers that were used as building blocks for the present manuscript. Topic 1: WHO 2021 classification of CNS tumors 1 Publication of a new WHO CNS tumor classification may in itself not be perceived as a major scientific achievement. However, quite some of the changes implemented in the fifth edition of the WHO Classification of Tumors of the Central Nervous System (WHO CNS5 classification) are the result of very good science, followed by (hopefully) ‘smart translation’ of the findings into a new classification, which definitely has a major impact on clinical neurooncology. Since November 2021, the WHO CNS5 classification is available online via https://tumourclassification.iarc.who.int, and the printed ‘Blue Book’ version can now be purchased as well (Figure 1). The review by Louis DN et al. published in Neuro-Oncology provides a very informative summary of major general changes in the WHO CNS5 classification and of specific changes in the different taxonomic categories 1.
Figure 1. Covers of the WHO CNS tumor Blue Books throughout the years.In these Blue Books, the actual WHO CNS tumor classification is the list of tumors that are recognized as distinct tumor types (see e.g. Table 1 for the 5th edition); the rest of the information in these books concerns explanation of the pathological (nowadays histological and molecular) characteristics of these tumors, and the most salient information on their clinical and radiological context. Molecular characteristics were for the first time introduced as defining criteria in the revised 4th edition, especially for diffuse gliomas in adults and for some embryonal tumors. In the 5th edition, molecular features are introduced as essential diagnostic criteria for many more tumors. In line with recommendations of the cIMPACT-NOW consortium 15, salient changes in this new classification are the separation of (groups of) pediatric-type low-grade and pediatric-type high-grade diffuse gliomas from adult-type diffuse gliomas, further refinement of the classification of ependymal tumors, and the addition of a few newly recognized tumors in the category of CNS embryonal tumors. Table 1 provides an overview of these changes in the WHO CNS5 classification (with # and * indicating the newly introduced and the provisionally accepted tumor types, respectively). Another important change is that grading of neoplasms is performed in the WHO CNS5 classification within (rather than across) tumor types, with Arabic (instead of Roman) numerals now used for the different grades. Thus, in the WHO CNS5 classification there is only one entry for e.g. astrocytoma, IDH-mutant, and one for meningioma (rather than separate entries for e.g. anaplastic astrocytoma, IDH-mutant, or for atypical meningioma). Also, glioblastoma, IDH-mutant should now be diagnosed as astrocytoma, IDH-mutant, CNS WHO grade 4.
Table 1. WHO classification of primary CNS tumors, 5th edition (2021).Major changes in the WHO CNS5 classification are the separation of (low-grade and high-grade) pediatric-type diffuse gliomas from adult-type diffuse gliomas; the recognition of several new ependymal, embryonal and other tumor types; assigning of CNS WHO grades within tumor types; coining the most malignant IDH-mutant diffuse astrocytic tumor as astrocytoma, IDH-mutant, CNS WHO grade 4; and changing the name of diffuse midline glioma (DMG), H3 K27M-mutant into DMG, H3K27-altered. See summary of Louis DN et al. 1 or, even better, the (digital version of the) ‘Blue Book’ for much more information of these changes. In RED & CAPITALS & ITALICS: overarching categories of tumors; in Bold and italics: groups of tumor types, and under these groups (with a few exceptions) the individual tumor types; # = newly defined tumor type compared to the 2016 edition; * = provisional tumor type; @ = tumor with revised nomenclature or revised placement. Click here to view a large version of this table. Furthermore, the presence of TERT promoter mutation, EGFR amplification and/or the combination of gain of complete chromosome 7 and loss of complete chromosome 10 can now be used for diagnosing an adult-type, histologically lower grade, IDH-wildtype diffuse glioma as glioblastoma, IDH-wildtype (CNS WHO grade 4), and the presence of homozygous CDKN2A/B loss to diagnose a histologically lower grade, IDH-mutant diffuse astrocytoma as CNS WHO grade 4. For yet other tumors the name or their placement in the classification was changed as well (@ in Table 1). For example, H3K27M-mutant after diffuse midline glioma (DMG) was changed into H3K27-altered because there are H3-wildtype DMGs especially in children that do show loss of nuclear H3K27me3 staining and with a similar prognosis as DMGs, H3K27M-mutant. Also, the name RELA fusion-positive for a subset of supratentorial ependymoma was changed into ZFTA fusion-positive, as ZFTA (‘zinc finger translocation associated’, the new name for c11orf95) appears to be the more frequent fusion partner in these tumors (most frequently showing fusion with RELA). Compared to the revised fourth edition, the WHO CNS5 tumor classification certainly is an improvement. However, as discussed in somewhat more detail in the last part of this review, this new classification brings several challenges as well, e.g., related to the (lack of) availability of molecular diagnostic tools, finding the optimal therapeutic management for newly defined tumor types, and the fact that a more refined taxonomy of CNS tumors makes it more difficult to perform studies on a large number of patients. Topic 2: Glioma driver mutations in the normal human brain 2 Cancer is a disease characterized by the accumulation of genomic aberrations that confer a proliferative advantage. Also, somatic mutations and copy number variations have been described to accrue with age in (seemingly) normal tissues 16. However, relatively little is known about this phenomenon in the normal brain. Ganz J and Maury EA et al. (with Lee EA and Walsh CA as corresponding authors) published a paper in Cancer Discovery 2 in which they used targeted gene sequencing on cerebral gray and white matter samples of 110 individuals to investigate the presence of oncogenic variants in normal brain tissue. The 121-gene panel was selected for genes implicated in different diseases and previously characterized oncogenic drivers. The authors hypothesized that somatic mutations would accumulate in brain tissue over time and (as neurons have a low proliferation capacity) would be enriched in cerebral white matter. The authors identified 35 variants present in normal brain tissue: 12 of the variants were in known proto-oncogenes or tumor suppressor genes, and 6 were known driver mutations of gliomas (IDH1, PTPN11, PTEN, NF1, APC, MTOR). The authors validated the variant allele frequencies (VAFs) from their gene panel with ultra-deep Ion Torrent sequencing. The most frequently found mutation was IDH1 R132H. In one patient, two adjacent white matter samples had vastly different variant allele frequencies (VAFs) of IDH1 R132H (5% vs 0.9%). Because a 5% VAF is high for a localized oncogenic mutation, the authors suggest that in this tissue, the mutation should be considered as a clonal event that conferred cells with a proliferative advantage. Using single-nucleus RNAseq, they found that the IDH1 R132H mutation was enriched in glial cells. Interestingly (and thus in contrast to what has previously been described for other organs), in this study the accumulation of oncogenic mutations in brain tissue did not positively correlate with age. In fact, all of the patients with identified somatic variants were under 30. Furthermore, in this study oncogenic mutations were found to be present in 5.4% of the non-diseased human brains, while the incidence of primary brain tumors in the population is much lower. These results thus raise some intriguing next questions, such as: What are the mechanisms for postnatal elimination of the mutant clones? What is the ‘tipping point’ that eventually causes brain tumor formation? Maybe the acquisition of a second hit (such as TP53 and/or ATRX mutation for IDH-mutant astrocytomas, and 1p/19q codeletion for IDH-mutant oligodendrogliomas) 17? And do maybe these findings help to explain the occurrence of ‘dual genotype’ oligoastrocytomas with histologically distinct astrocytic and oligodendroglial areas 18? Topic 3: Neuronal activity-dependent initiation of Nf1-mutant ‘optic gliomas’ 3 Over the past few years, we have begun to appreciate the unique role of neurons for glioma growth. For example, two papers that were already summarized in Topic 7 of the ‘Top ten discoveries in 2019’ review in this journal demonstrated the presence of bona fide neuron-glioma synapses in high-grade gliomas, with electrochemical-induced depolarization of the glioma membrane promoting proliferation of the tumor cells 19,20. These discoveries have boosted the interdisciplinary field of ‘cancer neuroscience’ 21. Therapies that target the neuron-glioma interface, especially through neuroligin-3 signaling (NLGN3), are now under investigation as a possible treatment for these malignant gliomas 22,23. In 2021, Pan Y et al. (with Monje M and Gutmann DH as corresponding authors) published a study in Nature on the impact of neuronal activity in the optic nerve on the formation and growth of ‘optic gliomas’ in mice with Nf1 mutation 3. The authors started with an authenticated genetically engineered mouse model which mimics the neurofibromatosis type 1 (NF1) childhood predisposition syndrome: a germline Nf1 mutation (Nf1+/-) combined with an acquired somatic Nf1 mutation in neural progenitor cells (Nf1-/-). When these mice are 9 weeks of age, gliomas form along the optic nerve, resembling what is seen in children with NF1 (and in the vocabulary of the WHO CNS tumor classification then generally concern pilocytic astrocytomas). By stimulating the retinal ganglion cells, the authors found an increase in optic nerve volume and proliferation rate, proving that optic nerve activity can increase optic glioma growth. Next, the authors investigated the effect of visual experience on the initiation and growth of these optic gliomas. Rearing mice in the dark from 6 weeks of age (before optic glioma formation) prevented the formation of tumors. Additionally, compared with mice raised in a regular light cycle, dark-rearing mice rescued retinal ganglion cell death and prevented optic nerve damage. Pan Y et al. further characterized the role of NLGN3, a synaptic adhesion molecule, in these models and found that it operates under a similar mechanism as in high-grade gliomas: ADAM10 sheddase cleaves and releases NLGN3 into in the tumor microenvironment, which stimulates proliferation of optic glioma cells. When the authors introduced a brain penetrant inhibitor of ADAM10 in their model, no tumor formed in the optic nerve. These insights raise many interesting follow up questions. For example, would limiting light exposure—e.g., with light-blocking glasses—and/or interference with NLGN3 during certain developmental periods in children help to prevent optic glioma formation? Hopefully, these findings will sooner or later provide new opportunities for the therapeutic management of children with NF1. Topic 4: Oncogenic role of H3 mutations in histogenesis context 4,5 Mutations affecting histone H3 are a defining feature in a particular subset of high-grade diffuse gliomas in children and suggest an epigenetic driver of cancer. The two more common mutations, H3.3 p.K28M (K27M) and H3.3 p.G35R/V (G34R/V), are mutually exclusive and typically lead to tumor formation in different CNS locations (H3 K27M in midline structures, and H3 G34R/V in the cerebral hemisphere). Two recent studies aimed to discern the oncogenic mechanisms of these H3.3 mutations using stem cell models of brain development and gliomagenesis. Haag D et al. (with Wernig M and Pfister SM as senior authors) published a study in Cancer Cell in which they investigated the effects of the H3 K27M mutation, which co-occurs in diffuse midline gliomas (DMGs) with a mutation in TP53 in 77% of cases 4. Previous single cell RNAseq studies of DMGs, H3 K27M-mutant revealed that these tumors may originate from an oligodendrocyte progenitor cell (OPC) 24. Haag et al. inserted a K27M mutation in neural stem cell (NSC) and OPC lines, which increased proliferation in these cell lines. Interestingly, only NSC lines with K27M and knock-out of TP53 formed malignant tumors in mouse xenografts that resembled the pathology of DMGs. As this finding is inconsistent with prior reports of the cell-of-origin, the authors hypothesized that the K27M mutation could cause NSC to adopt an OPC-like transcriptional profile. To this end, the authors used chromatin immunoprecipitation sequencing (ChIP-seq) to describe the interactions between mutant H3.3 and DNA. They found H3.3 enrichment in bivalent chromatin domains which maintains the expression genes necessary for pluripotency and stem cell identity. This can explain how K27M-mutant NSCs can mimic OPC transcriptional programs and could drive these tumors. Bressan RB et al. (with Pollard SM as lead contact) described the oncogenic effects of the G34R mutation in NSC lines derived from the forebrain and hindbrain in a paper in Cell Stem Cell 5. Forebrain NSC lines with only the G34R mutation had low tumorigenic capacities. However, combination of knockout of TP53 and amplification of PDGFRA in these cells resulted in increased proliferation and the formation of malignant tumors when xenografted in mice. The authors then sought to describe the epigenetic effects of the H3 G34R mutation. Unlike the K27M mutation, which causes wide-spread epigenetic and transcriptional changes, the authors found that the G34R mutation reduces binding with ZMYND11, a tumor suppressor that alters the elongation and splicing of highly expressed genes. In the context of brain tumors, this results in higher expression of genes that regulate forebrain development and locks NSCs into a proliferative and non-differentiated state. Together, these papers very nicely show how pre-clinical stem cell modeling can untangle the mechanisms of cancer initiation in general, and of the oncogenesis of H3.3 mutant tumors in particular. Topic 5: Epigenomic insights into glioma cell differentiation and plasticity 6,7 Single-cell (sc) RNAseq studies of diffuse gliomas have revealed unprecedented insights into their intra-tumoral heterogeneity, wherein glioma cells from the same tumor exist in different transcriptionally defined cell states 25-27. Many of these cell states mirror the neurodevelopmental hierarchy, and it has been suggested that tumor cells can transition between more or less differentiated states. Such a plasticity may well contribute to resistance to therapy. As gene regulation and DNA methylation (DNAme) can largely control these cell states, back-to-back papers published in Nature Genetics by Johnson KC and Anderson KJ et al. (with Verhaak RGW as senior author) 6 and Chaligne R, Gaiti F, Silverbush D, and Schiffman JS et al. (supervised by Suvá ML and Landau DA) 7 measured DNAme and RNA expression at the single-cell level of IDH-wildtype glioblastomas in adults (IDH-wt GBMs) and in IDH-mutant (IDH-mt) diffuse gliomas. Using scDNAme, both articles report on how glioma cells from the same patient exist in different methylation states, while cells from different patients can exist in the same states. These studies re-affirm that diffuse gliomas, especially GBMs, have high intra-tumoral heterogeneity, with malignant cells adopting different epigenotypes. Obviously, the bulk methylation assays that are frequently used to classify (glial) brain tumors fail to account for this level of intra-tumoral heterogeneity that is uncovered by such single-cell analyses. Both articles took different approaches to describe the epigenomic landscape that gives rise to the differentiation hierarchies in gliomas. Chaligne et al. report that, compared to IDH-mt gliomas, Polycomb complexes in IDH-wt GBMs are more hypermethylated in stem-like cells than in differentiated cells. This suggests a role for DNAme in maintaining a stem-like phenotype of GBM cells, thereby contributing to their treatment resistance and progression. However, this was not seen in IDH-mt gliomas, suggesting a different role of DNAme in maintaining their stem-like cell states. Furthermore, the authors constructed lineage trees using scDNAme and found that cells within each clade had the same chromosomal aberrations. This highlights how sub-clonal mutations are related with the epigenotype of glioma cells. Interestingly, when the authors inferred glioma cell state (NPC, OPC, AC, MES) based on scRNAseq for IDH-wt GBMs, they found that cells within the same lineage adopted different transcriptional states. However, the reverse was found for IDH-mt glioma, where closely related cells obtained similar cell states. Altogether, these results confirm that IDH-wt GBMs have higher cell plasticity than IDH-mt glioma, and DNAme contributes to the glioma stem-like identity. Johnson et al. also investigated the contribution of ‘DNAme disorder’ (i.e., aberrant methylation which allows cells to adapt to diverse methylation states) for maintaining the stem-like identity in gliomas. DNAme disorder could allow for the glioma cells to overcome environmental stressors and contribute to resistance to treatment. This hypothesis was tested in vitro by exposing two glioma cell lines to hypoxic conditions and irradiation. The authors found that under such environmental stressors, DNAme disorder increases with time, potentially allowing the cells to adopt and retain their stem-like identities. Topic 6: Proteogenomic and metabolomic characterization of glioblastoma 8 Wang L-B et al. (with the Clinical Proteomic Tumor Analysis Consortium) published a study which integrated whole exome/whole genome sequencing, bulk and single-nucleus RNA sequencing (RNAseq), DNA methylation, proteome, phospho-proteome, acetylome, lipidome and metabolome datasets for 100 treatment-naïve glioblastomas. Landmark studies previously already comprehensively characterized the genome and transcriptome of glioblastoma (see e.g. 28). A deep ‘multi-omic’ approach, like Wang L-B et al. undertook, provides very interesting further insights into functional and potentially targetable differences between glioblastoma subtypes. Based on their findings, especially with regard to gene expression, and protein and phosphoprotein abundances, Wang L-B et al. defined three ‘multi-omic subtypes’ which closely resemble TCGA (The Cancer Genome Atlas) glioma subtypes: the proneural-like subtype was enriched for neurotransmission and synaptic vesicle gene-sets; the mesenchymal-like subtype was enriched for immune system activation, phagocytosis, and glycolysis; finally, the classical-like subtype was enriched for chromatin modification, DNA repair, and mRNA splicing. The authors also defined immune-based subtypes based on gene-set enrichment analysis of RNAseq data. Subtypes ranged from low-enrichment of all immune cells (especially classical and proneural-like glioblastomas) to enrichment of lymphocytes, microglia, and macrophages (mesenchymal-like glioblastomas). The authors integrated these observations with single nucleus RNAseq of 18 samples spanning all multi-omic and immune subtypes, and characterized expression programs found in the neoplastic, stromal, and immune cells. Interestingly, this RNAseq analysis revealed upregulation of epithelial-mesenchymal transition related genes in both glioma and immune cells of mesenchymal-like glioblastomas. These differences could be observed on the protein-level as well. Last but not least, the authors also noticed subtype-specific enrichment of lipids and metabolites. For example, they noticed IDH mutation status-associated differences in glycolysis-related metabolites. Not surprisingly, IDH-mutant gliomas showed increased levels of 2-HG, as well as decreased glutamate and serine. Phosphoproteomic data highlighted therapeutic opportunities for treating glioblastoma, with PLCG1 and PTPN11 as signaling hubs for receptor tyrosine kinases. Importantly, all the data published by Wang et al. is open access and can thus be used as a resource for future studies of glioblastomas. Indeed, such a rich dataset adds depth to the previous knowledge from genomic and transcriptomic investigations of glioblastomas and can hopefully pave the way for more effective, personalized treatments for these tumors. Topic 7: Genomic and immunologic heterogeneity of gliomas and brain metastases 9 Immunotherapies for primary and secondary brain tumors are a ‘hot topic’ in the field of neurooncology (see for example also Topic 9 of the ‘Top ten discoveries of the year 2019’ review in this journal). In 2021, Schaettler MO and Richters MM et al. (with Griffith M and Dunn GP as supervising authors) published a study in Cancer Discovery investigating intra-tumoral heterogeneity of somatic variants, neo-antigens, and infiltrating T-cells in gliomas as well as in brain metastases. For this study, the authors collected 93 spatially separated tumor samples from 30 patients. These samples were subjected to whole exome, RNA and T-cell Receptor sequencing (WES, RNAseq, TCR-seq). Interestingly, while brain metastases had considerably more somatic variants per tumor than gliomas, these variants were mostly clonal, so spanning all tumor samples of a patient. When analyzing the sub-clonal variants of gliomas, the authors noted that sequencing only one region of the tumor missed 40% of variants that were obtained when sequencing three regions. Additionally, they found that 9/16 gliomas in this study showed characteristics of multiple transcriptional subtypes (classical, mesenchymal, neural and/or proneural). Integrating WES and RNAseq data allowed the authors to predict HLA class I and II neo-antigens expressed in the tumors. They found that the neo-antigens in brain metastases were more often clonal. This could have implications for the development of personalized immunotherapies, as clonal neoantigens would be better targets. Using RNAseq data, the authors also described the immune composition of the tumors. Brain metastases had robust infiltration of monocyte-derived macrophages, while gliomas were more enriched for microglia gene signatures. Finally, the results of TCR-seq analysis revealed that spatially distinct samples of brain metastases had more similar T-cell clones than those of gliomas. Altogether, these findings highlight the considerable intra-tumoral heterogeneity in somatic variants, transcriptome, and neo-antigens of gliomas. This can be expected to pose significant challenges for developing effective immunotherapies. Comparatively, brain metastases are more homogenous tumors, which could partially explain the clinical benefits of immune checkpoint inhibition when treating patients with those latter tumors. Topic 8: Therapeutic interference with glioblastoma cell membranes using Prozac® 10 So far, little progress has been made with improving the prognosis for patients with IDH-wildtype glioblastomas (IDH-wt GBMs). GBMs are known to have an altered lipid composition, with molecular alterations being linked to differential plasma membrane remodeling 29. For example, GBMs are frequently driven by the amplification of EGFR on extra-chromosomal DNA (ecDNA). Furthermore, variants in the extracellular EGFR domain (especially EGFRvIII) cause constitutive activation 30,31. These signaling molecules are organized into lipid rafts on the plasma membrane. In a study of Bi J et al. published in Cell Reports (with Mischel PS as lead contact), the authors identified that GBMs are dependent on sphingomyelin phosphodiesterase 1 (SMPD1), an enzyme that converts sphingomyelin to ceramide and alters the composition of the plasma membrane. In the TCGA (The Cancer Genome Atlas) cohort of GBM samples, increased expression of SMPD1 was associated with shorter overall survival, suggesting that targeting this pathway could be a therapeutic strategy for GBM. Interestingly, fluoxetine (sold under the brand name Prozac®; a brain-penetrant, FDA-approved selective serotonin reuptake inhibitor (SSRI) used to treat multiple psychiatric disorders) inhibits SMPD1 activity. In vitro experiments of GBM patient-derived cell lines treated with fluoxetine was found to result in an increase in lysosomal stress and dose-dependent cell death. Also, temozolomide showed synergistic effects with fluoxetine. These results were confirmed in in vivo studies of GBM orthotopic xenografts, wherein 6 of 8 mice receiving both temozolomide and fluoxetine showed no tumor recurrence after 5 months of treatment. Over-expression of SMPD1 in vitro and in vivo reversed the effects of fluoxetine. Next, the authors examined electronic medical records from 180 million Americans who were diagnosed with GBM between 2003 and 2017. The patients with GBM who, for psychiatric reasons, were also treated with fluoxetine (Prozac®) during the disease course of their brain tumor had significantly longer overall survival. This result was not found for GBM patients treated with other SSRIs. In conclusion, the results of the study of Bi et al. indicate that fluoxetine/Prozac® can maybe be ‘re-purposed’ and integrated with standard of care of patients diagnosed with GBM in order to help improve their survival. Obviously, controlled clinical trials are necessary to substantiate this idea further. Also, it would be interesting to learn more about other compounds that might be good candidates for therapeutic interference with (the composition of) GBM cell membranes. Topic 9: Liquid biopsy assessment of residual medulloblastoma 11 A current problem in the management of children with medulloblastoma is assessing residual disease during treatment and predicting which patients relapse locally or through dissemination to the leptomeninges (which, unfortunately, frequently leads to death) 32. Currently, MRI and cytology assessment of the cerebrospinal fluid (CSF) are used to detect relapse. However, these diagnostic modalities are generally only useful once the tumor has progressed substantially. Medulloblastomas typically have few driver mutations and are characterized by high levels of chromosomal instability (copy number variants). In a study from 2020, Escudero et al. found that CSF-derived cell-free DNA (cfDNA) recapitulated molecular characteristics of the initial medulloblastomas, including the oncogenic drivers 33. cfDNA from the CSF could also be used to classify medulloblastomas into the four molecular sub-groups. Following up on these findings, Liu API, Smith KS, and Kumar R et al. (with Gajjar A, Robinson GW and Northcott PA as senior authors) published a paper in Cancer Cell evaluating the utility of cfDNA to assess measurable residual disease (MRD) for guiding the therapeutic management of medulloblastoma patients 11. They performed low-coverage whole genome sequencing on cfDNA from 476 CSF samples, representing 123 patients, and derived CNVs as a biomarker for MRD. Of the 105 CSF samples collected at baseline, the authors detected CNVs in 64% of the samples. CNVs were not detected in cfDNA from CSF of patients without oncological diseases that were used as control. Using multivariate regression, they found that detection of baseline samples was associated with tumor location and subgroup, wherein detection was lowest for tumors centered within cerebellar hemispheres or vermis, and for the SHH subgroup. Additionally, the detection of MRD in samples post-radiotherapy, mid-chemotherapy, or at the end of therapy was associated with worse progression free survival (PFS), while baseline CNV detection was not prognostic for PFS. The authors did find a high correlation between CNVs detected at baseline in the CSF and in the primary tumor, confirming the results from Escudero et al. Comparing serial samples of cfDNA, 75% of patients had a more ‘unstable’ genome with a loss of chromosome 10q at recurrence. MRD was detected for all patients with persistent disease recurrence, demonstrating the high accuracy for this assay. Of note, of the patients with medulloblastoma recurrence, MRD was detected by CSF cfDNA analysis at least 3 months before appearing on MRI or based on cytology analysis. This study demonstrates that detection of CNVs from CSF-derived cfDNA may allow for a more sensitive evaluation of disease progression in children with medulloblastoma than the currently available tools. The authors therefore advocate for incorporating such cfDNA analysis of liquid biopsies to inform management of these aggressive cancers. Topic 10: Improved (histo)molecular grading of meningiomas 12-14 Until recently, the diagnosis WHO grade II meningiomas was based on the presence of increased mitotic activity, brain invasion, presence/absence of three or more of the following features: high cellularity, small cells with a high nucleus: cytoplasm ratio, prominent nucleoli, patternless/sheet-like growth, and foci of necrosis. Furthermore, particular histological phenotypes were used to assign a WHO grade II (clear cell, chordoid) or WHO grade III (rhabdoid, papillary) to meningiomas, while very high mitotic activity and/or overtly malignant cytomorphology were considered as sufficient for the diagnosis of malignant/anaplastic meningioma (WHO grade III) as well. However, the prognostic meaning of this histology-based grading system was suboptimal, and it became increasingly clear that molecular data can be used to refine grading of these tumors. Indeed, in the WHO CNS5 classification, presence of TERT promoter mutation and/or homozygous CDKN2A and/or CDKN2B deletion are now listed as criteria for meningioma, CNS WHO grade 3. Furthermore, in this new classification, the rhabdoid and papillary phenotypes are no longer considered as CNS WHO grade 3 based on their histological phenotype alone 1,34. Nassiri F et al. (with Aldape K and Zadeh G as supervising authors) published a paper in Nature providing a wealth of matched multidimensional bulk and single-cell molecular and clinical data on a large cohort of meningiomas, enriched for the higher-grade tumors according to WHO 2016 criteria 12. Unsupervised sample-wise clustering of gene-level somatic copy-number alterations (CNAs), DNA methylome, and transcriptome data in isolation revealed six stable subgroups for each datatype with clinically relevant and significant differences in outcome. Additional (second-order) clustering revealed four stable molecular groups (MG1–MG4) without a clear one-to-one relationship between molecular group and WHO grade. Classification by molecular groups was independently associated with recurrence-free survival as assessed by multivariable Cox regression, even after accounting for known prognostic clinical factors. MG3 and MG4 tumors (carrying the most unfavorable outcomes) were found to be high-aneuploidy tumors with losses in chromosomes 22q, 1p, 6q, 14, and 18. In addition, MG4 meningiomas showed a gain of chromosome 1q and a loss of chromosome 10. Such findings have the potential to supersede existing molecular and clinically used classifications and grading schemes. Indeed, two studies published in 2021 show that prognostication for patients with meningioma can be improved by taking particular copy number variations (CNVs) into account (Figure 2). Driver J et al. (corresponding authors Bi WL and Santagata S) published a paper in Neuro-Oncology evaluating whether the use of chromosomal copy-number data provided more accurate prediction of time to recurrence for patients with meningioma than the traditional WHO grades 14. Their discovery cohort consisted of 527 patients diagnosed with meningioma, and two independent cohorts of 172 meningioma patients were used for validation of the findings. Based on mitotic count and the presence/absence of the loss of (arms of) particular chromosomes (1p, 3p, 4, 6, 10, 14q, 18, 19), and of homozygous deletion of CDKN2A the authors developed a scheme with three Integrated Grades (1-3). This grading approach was found to more accurately identify meningioma patients at risk for recurrence than the traditional WHO grading system.
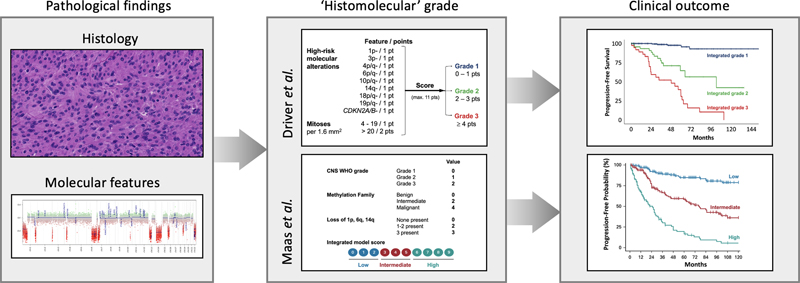
Figure 2. Assessment of chromosomal copy number aberrations for improved grading of meningiomas.Both in the study by Driver J et al. 14 and by Maas SLN et al. 13 the presence/absence of particular chromosomal losses is used for assessment of prognosis of meningiomas. In the ‘Driver approach’, this is combined with information on CDKN2A/B status and number of mitoses, while in the ‘Maas approach’ the grade as traditionally assigned based on histological features and the suggested methylation family using the meningioma classifier (benign – intermediate – malignant) are taken into account as well. Importantly, next to methylation array-based grading system for meningiomas, the study of Maas et al. presents some alternatives for a more stepwise approach for grading of meningiomas, taking into account that particular histological phenotypes (angiomatous, psammomatous, secretory) are strongly associated with CNS WHO grade 1 behavior, while presence of TERT promoter mutation and/or CDKN2A/B loss indicate high-risk tumors. Maas SLN, Stichel D, Hielscher T, Sievers P et al. (supervisors von Deimling A and Sahm F) published a study in the Journal Clinical Oncology in which DNA methylation profiling and copy-number information were generated for 3,031 meningiomas of 2,868 patients and mutation data for 858 samples 13. Both CNV- and methylation family-based subgrouping independently resulted in increased prediction accuracy of risk of recurrence compared with WHO grading. Prediction power for outcome was assessed in a retrospective cohort of 514 patients, and validated on a retrospective cohort of 184 as well as on a prospective cohort of 287 cases. Combining different risk stratification approaches into an integrated molecular-morphologic score resulted in substantial further increase in accuracy. Again (like in the study of Driver et al.), the integrated scores were found to separate tumors more precisely for risk of progression, especially so at the diagnostically challenging interface of CNS WHO grade 1 and grade 2 tumors. Discussion In 2021 again an amazing amount of information with relevance for neurooncological pathology has been published. Hopefully, this review helps readers keep up with what’s new in this respect. Like WHO tumor classifications in general, and as already stated in the section on Topic 1, the WHO CNS5 classification represents work in progress with room for further improvements. Obviously, this new classification brings further challenges as well. For example, for more CNS tumors it is now impossible to reach a state-of-the-art ‘histomolecular’ diagnosis in case molecular tools for assessment of essential diagnostic characteristics (or immunohistochemistry for reliable surrogate markers, see below) are not available. In those situations, adding NOS (not otherwise specified) to the histology-based diagnosis is the way to go 35. Furthermore, designing the optimal therapeutic management for newly defined tumor types is challenging. And while a more precise classification facilitates enrollment of more homogeneous populations of patients in clinical studies, the higher granularity of CNS tumor taxonomy makes it more difficult to perform studies on a large number of patients for particular tumor types. Still, one would like to think that patients suffering from a CNS tumor are better served by a more precise diagnosis because this allows for a better estimation of prognosis and, hopefully sooner than later, for a more tailored and effective therapeutic approach. Immunohistochemistry for surrogate markers can indeed provide a way for making a bona fide, ‘histomolecular’ WHO CNS5 diagnosis. Some examples of stains that are already often used in clinical practice are immunohistochemistry for mutant proteins (IDH1 R132H, H3 K27M, H3 G34V/R, BRAF V600E), for loss of staining for normal proteins in the tumor cell nuclei (ATRX, INI1, BRG1, H3K27me3), and for abnormal location or intensity of a protein in the tumor cell nuclei (e.g. STAT6, p53). The information provided by the studies of Driver et al. and of Maas et al. (see Topic 10) shows that the care for patients with meningioma can be improved by assessment of particular molecular characteristics for the grading of these tumors 13,14. At the same time, there may be an opportunity for immunohistochemistry here as well: for example, loss of H3K27me3 staining of tumor cell nuclei in meningioma has been reported as an additional tool for identification of meningiomas with higher risk of recurrence 36,37. Furthermore, in the study of Nassiri F et al. (see Topic 10), particular proteins were found to be highly enriched in the different molecular groups (MG1: S100B; MG2: SCGN; MG3: ACADL; MG4: MCM2) 12. Further validation of such immunohistochemical approaches and comparison with the results as presented by Driver et al. and by Maas et al. is needed. Also, acknowledging that the molecular underpinnings in meningiomas diagnosed in kids are distinct from those in adults 38, pediatric meningiomas may require an adapted grading system. While CNS tumors can now be much more precisely characterized than a few decades ago, the translation of this increased knowledge into more effective treatments is seriously lagging behind. Quite some of the Topics discussed in this review concern studies that further elucidate the pathobiology of particular CNS tumors. Hopefully, such knowledge can be exploited for more effective therapies as well, e.g., by targeting of epigenetic regulation of neural or oligodendrocyte precursor states/cells (Topic 4), by remodeling the epigenome of glioma cells in order to alter the differentiation hierarchy of gliomas (Topic 5), by exploiting the new information obtained by multi-omic investigations for improved targeting of the pathways involved in glioblastomas (Topic 6), and/or by improving strategies for selection of patients for targeted (immune)therapies based on immunophenotyping studies and information on the spatial distribution of genomic alterations of gliomas/glioblastomas (Topic 7). In a study of > 10,000 cancer patients across 20 different cancer types, transcriptomic analysis allowed for the identification of four distinct tumor-microenvironment subtypes which correlated with patient response to immunotherapy. By integrating transcriptomic and genomic data, a global tumor portrait can be designed, describing the tumor framework, mutational load, immune composition, anti-tumor immunity, and immunosuppressive escape mechanisms, guiding therapeutic decision-making 39. The finding that visual experience and neuronal activity is required for the formation and growth of optic gliomas may provide new targets for therapeutic interference for these tumors (Topic 3), and maybe some ‘old drugs’ can indeed be re-purposed in order to improve the outcome for patients with glioblastoma, e.g. because they interfere with the cell membranes of the tumor cells (Topic 8). Interestingly, a recent (‘seed & soil’) study on the metastatic potential of 500 human cancer cell lines spanning 21 types of solid tumor, breast cancer cells capable of metastasizing to the brain were found to have an altered lipid metabolism, perturbation of which indeed resulting in curbed development of brain metastasis 40. As discussed under Topic 9, a liquid biopsy approach using detection of CNVs in CSF-derived cfDNA may allow for improved recognition of disease progression in children with medulloblastoma. However, in a very recent study of 258 pediatric brain tumors ‘across all histopathologies’, CNVs were detected in only 20% of CSF-derived cfDNA, and the fact that no genomic aberrations could be detected in liquid biopsies from patients with low-grade gliomas may indicate that its utility is more promising for patients with aggressive brain tumors 41. Papers published in 2021 that the authors considered as (very) interesting, but that didn’t make it to the Top ten list include: publications presenting potential new tumor types (e.g., supratentorial neuroepithelial tumors, PLAGL1 fusion-positive 42 and histologically polyphenotypic neuroepithelial tumors, PATZ1 fusion-positive 43); the report of detailed molecular analysis of (plus clinical variables in) a series of 191 medulloblastomas in adults 44; a study reporting that germline variants in the E-cadherin gene CDH1 are (in addition to non-CNS tumors, especially of the stomach and breast) associated with increased risk of neuroepithelial tumors/oligodendrogliomas, IDH-mutant, 1p/19q-codeleted 45; papers reporting the accuracy of different molecular tests for assessment 1p/19q status 46, and for MGMT promoter methylation status 47,48. Also, for those with a keen interest in neuroimmunooncology it may be good to know that dural sinuses appear to act as a ‘neuroimmune interface’ where brain antigens are surveyed, and that may show age- and disease-related dysfunction 50, and that bone marrow niches in the skull and vertebral column act as myeloid cell reservoirs for the meninges and CNS parenchyma 51. Acknowledging that for multiple decades histological slides formed the basis for the diagnosis of (CNS) tumors, one can argue that for a long time clinical (neuro)pathologists have acted as masters of ‘thin slicing’ (i.e., making quick inferences about characteristics of a tumor using a limited amount of tissue). Nowadays, however, for the pathological diagnosis of a rapidly increasing number of CNS tumors assessment of particular molecular characteristics is required as well. And in the near future, spatial and/or multi-omics analysis of these tumors may also be required. One can expect that by that time, diagnostic support tools will become available, such as tools for Artificial Intelligence (AI)-guided analysis of digitized, histological slides 52, and visualization tools for integration of the multi-omic profiling data and for guidance of therapeutic decision-making 39 (Figure 3). While such a transition from thin slicing towards augmented reality indicates that the future of neurooncological pathology is bright, it wouldn’t mean too much unless these developments indeed can help to drastically improve the prognosis of patients that suffer from a CNS tumor.
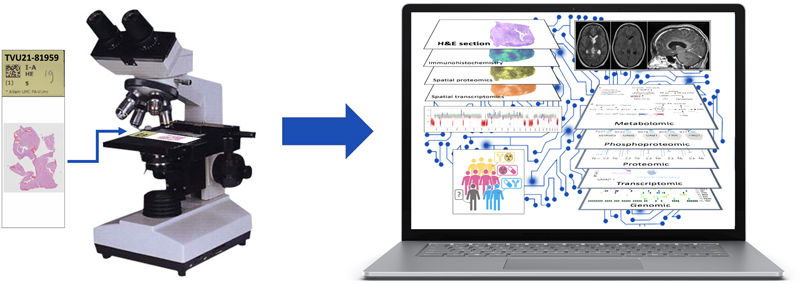
Figure 3. Neurooncological pathology: from thin slicing to augmented realityAs histological slides formed the basis for the diagnosis of (CNS) tumors for multiple decades, one can argue that for a long time clinical (neuro)pathologists have acted as masters of ‘thin slicing’. This term was initially used especially in psychology and philosophy for a situation in which one makes very quick inferences about the state, characteristics or details of an individual or situation with minimal amounts of information. In the context of tumor pathology, it could literally mean the ability to make quick inferences about characteristics of a tumor using very thin slices of tissue. Nowadays, however, for an increasing number of CNS tumors, molecular characterization is necessary as well. In the near future, spatial and/or multi-omics analysis of CNS tumors may be required for an optimal assessment of the diagnosis (including prognosis) and for prediction of the best therapeutic management. One can expect that by that time, diagnostic (AI-guided and visual) support tools have become available that guide the neurooncological pathologist from a more bounded rationality into an augmented reality. Meanwhile, it is important to realize that the judgement of an experienced (neuro)pathologist based on thin-slicing/histological slides will remain invaluable for an optimal diagnosis and indeed in quite some situations (e.g., tumors that can readily be recognized based on an H&E stained section +/- some additional immunohistochemical stains, and in case of problems with representativeness or a differential diagnosis with non-neoplastic lesions) can even be more accurate than judgements based on much more, non-histological information. References
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |