|
|
|
Free Neuropathology 3:9 (2022) |
|
Original Paper |
|
Soluble brain homogenates from diverse human and mouse sources preferentially seed diffuse Aβ plaque pathology when injected into newborn mouse hosts |
|
Brenda D. Moore1,2,3, Yona Levites1,2,3, Guilian Xu1,2,3, Hailey Hampton1,2, Munir F. Adamo1,2, Cara L. Croft1,2,3, Hunter S. Futch1,2, Corey Moran1,2, Susan Fromholt1,2, Christopher Janus1,2,3, Stefan Prokop2,3,4,5, Dennis Dickson6, Jada Lewis1,2,3, Benoit I. Giasson1,2,3, Todd E. Golde1,2,3,7, David R. Borchelt1,2,3 |
|
1 Department of Neuroscience, College of Medicine, University of Florida, Gainesville, FL 32610, USA |
|
Corresponding author: |
|
Submitted: 18 January 2022 Accepted: 28 February 2022 Copyedited by: Henry Robbert Published: 23 March 2022 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Amyloid-β, Tau, α-Synuclein, Seeding, Alzheimer’s disease, Lewy body dementia |
|
Abstract
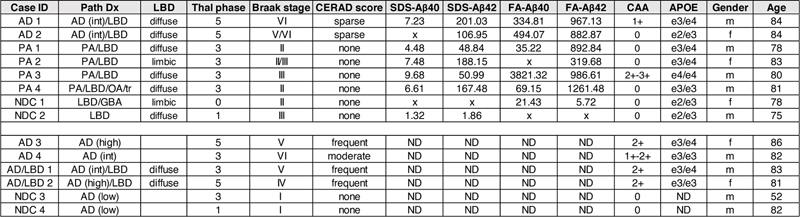
Background: Seeding of pathology related to Alzheimer’s disease (AD) and Lewy body disease (LBD) by tissue homogenates or purified protein aggregates in various model systems has revealed prion-like properties of these disorders. Typically, these homogenates are injected into adult mice stereotaxically. Injection of brain lysates into newborn mice represents an alternative approach of delivering seeds that could direct the evolution of amyloid-β (Aβ) pathology co-mixed with either tau or α-synuclein (αSyn) pathology in susceptible mouse models. Abbreviations AD - Alzheimer’s disease, LBD - Lewy body disease, AD/LBD - Alzheimer’s disease/Lewy body disease, Aβ - amyloid-β, αSyn - α-synuclein, APP - Amyloid precursor protein, PA - Pathological Aging, GFAP - Glial fibrillary acidic protein, MAPT - Microtubule-associated protein tau, NDC - Nondemented Controls, WT - Wild type, CAA - cerebral amyloid angiopathy, ELISA - Enzyme linked immunosorbent assay, SDS - Sodium dodecyl sulfate, FA - Formic acid, Thio-S - Thioflavin S. Background The amyloid cascade hypothesis posits that deposition of the Aβ peptide is an initiating factor in Alzheimer’s disease (AD) with downstream effects of increased inflammatory response, altered neuronal homeostasis, altered tau phosphorylation leading to tangle formation, neuronal dysfunction, and cognitive decline (1, 2). The interaction between Aβ and tau pathology has been modeled in transgenic animals that co-express human amyloid precursor proteins (APP) with mutations associated with familial AD (FAD) and wild-type or mutant human tau (3-12). Multiple studies report that the presence of Aβ pathology exacerbates the severity of tau pathology, leading to increased tau phosphorylation or increased levels of neurofibrillary tangles (3, 6, 8, 10, 11). Interactions between Aβ and tau pathology have also been modeled in mice by utilizing the prion-like capacity of Aβ and tau aggregates to seed accelerated AD pathology. Aβ containing seeds, derived from human AD and Pathological Aging (PA) brain lysates, have been shown to accelerate the time-course and severity of Aβ deposition in amyloid precursor protein (APP) transgenic models as well as impact the morphology of the seeded deposits (for review see (13)). Seeding preparations can consist of human brain homogenates, APP transgenic mouse brain homogenates, enriched preparations derived from such homogenates and synthetic Aβ fibrils (13). In most of these published studies, seeds are injected into adult APP mice that are a few months away from developing Aβ pathology on their own and in such cases the inherent pathological tendencies of the APP host model can contribute to the pathological outcome. Partially purified tau aggregates from AD brains were found to seed enhanced neuritic tau pathology in the 5xFAD mouse model and the AppNL-G-F/human MAPT double knock-in model (14); however, neither of these models produced pathology resembling neurofibrillary tangles. In APPswe/PS1dE9 and AppNL-F/NL-F mice crossed to humanized hTau mice, the presence of Aβ pathology did not produce significant enhancement of tau seeding (15). These latter studies illustrate the need to refine seeding approaches towards producing models that reproduce a broader spectrum of AD-related pathology. Recent studies have begun to use seeding approaches to create models of mixed AD or Lewy body disease (LBD) pathology. In Parkinson’s disease and LBD, the presence of Aβ plaques is associated with cortical αSyn pathology (16, 17). Similar to studies of Aβ/tau interactions, investigations of Aβ and αSyn interaction in transgenic mice have suggested a possible synergistic relationship (18, 19). In seeding models involving the injection of αSyn preformed fibrils, Aβ deposits accelerated αSyn pathogenesis and spread throughout the brain of 5xFAD mice (20) and in bigenic mice co-expressing APPswe/PS1dE9 and WT αSyn (21) . One of the goals of our study was to further explore the potential to use seeding as a means to generate novel mouse models of mixed AD and LBD pathology. A subset of cognitively normal aged individuals exhibit substantial Aβ pathology at autopsy (22). These individuals, which we use the designation of Pathologic Aging (PA), may represent prodromal, presymptomatic AD (23-26). The amyloid pathology of PA brains has typically been described as diffuse in nature whereas AD brains contain both diffuse and compact/neuritic deposits of Aβ (26-28). Cognitively normal individuals with PA generally have very low levels of tau pathology (26). Rigorous biochemical examination of Aβ from the pre-frontal cortex of PA and AD brain revealed extensive overlap in Aβ levels, peptide profiles, solubility, and SDS-stable oligomeric assemblies (25). Thus, PA brains provide a source of human brain-derived Aβ seeds that can be compared to human AD brain preparations that contain both Aβ and tau seeds. Nearly all of the published studies involving injection of Aβ, tau, or αSyn seeds into the brains of transgenic mice have followed a protocol of injecting the seeds stereotaxically into the brains of young adult mice. In this study, we have examined the efficacy of intracerebroventricular injection of homogenates prepared from AD, PA, and AD/LBD donors into newborn APP, APP/tau, or APP/αSyn host mice. In prior studies, we had established that injection of recombinant adeno-associated virus into newborn mice was an effective approach to obtaining widespread dispersion of injected virus (29, 30). We therefore hypothesized that misfolded seeds of Aβ, tau, or αSyn might similarly be more widely dispersed to more effectively seed pathology. In fact, we observed efficient seeding of αSyn pathology in newborn mice, expressing A53T human αSyn (M83 line), by injecting brain lysates from patients with multisystem atrophy (31). Here we performed brain lysate injection studies at P0 in the lateral ventricles of host transgenic mice that co-express humanized mutant APP with either mutant human tau-P301L or wild-type human αSyn (M20 line). Importantly, the recipient transgenic mice used in the current studies develop pathology at late ages or not at all; enabling sensitive evaluation of potential seeding. We observed that injecting newborn APP, APP/tau, or APP/αSyn mice with homogenates from diverse pathological specimens, including PA brains, produced robust diffuse Aβ deposition. Though the human homogenates used contained coexisting αSyn and tau seeds, we were not able to directly induce LBD or tau pathology in recipient models tested here. We compared the data from human lysates to lysates prepared from transgenic mouse donors finding again that diffuse Aβ deposits were preferentially seeded when these preparations were injected in newborn mice. APP mice seeded with AD brain lysates, exhibiting diffuse Aβ pathology, also showed no significant impairments in a contextual fear paradigm of cognitive performance. Our findings indicate that the injection of human brain homogenates from AD and AD/LBD donors into newborn APP mice preferentially induces diffuse Aβ pathology. Interestingly, PA brains induce the same pathology implying that AD and AD/LBD brains contain amyloid seeding entities that are shared with PA brains. Methods Description of AD, PA and NDC cases For this study we utilized a subset of the human brain samples that our group previously analyzed (25). Tissue sections and frozen pre-frontal cortex (AD = 2, PA = 4, and NDC = 2) were obtained from the Mayo Clinic Brain Bank with informed consent, in accordance with the Mayo Clinic institutional review board, using previously described acquisition and diagnostic analyses (26, 32, 33). Samples were initially cryo-pulverized to allow for multiple studies. We previously analyzed the Aβ peptide profile, quantity, and solubility in these brain samples (25). In this study, we analyzed two brains from AD patients (both aged 84), four PA brains from subjects (age range = 78 to 83) without clinical evidence of dementia, and two brains with rare or no AD lesions from elderly individuals without clinical evidence of a neurological illness (ages 75 and 78). In addition, human brain samples were also obtained from the University of Florida Neuromedicine Human Brain Tissue Bank following institutional regulations and previously described classification (34-36). We analyzed tissue sections and frozen pre-frontal cortex from two AD patients (ages 82 and 86), two cases with AD/LBD (ages 81 and 83), and two control individuals without pathology (ages 52 and 82). Table 1 summarizes the cases studied in this report. Table 1. List of human brain samples used in this study.
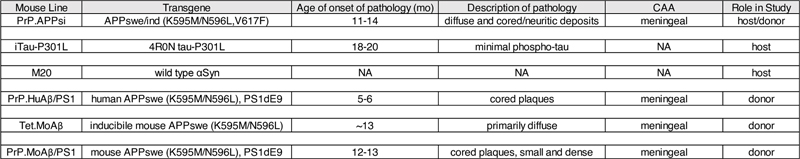
Pathological diagnosis (PathDx), Lewy body disease (LBD) classification, amyloid plaque score (Thal phase), Braak stage, Consortium to Establish a Registry for Alzheimer’s disease (CERAD) protocol, 2% SDS and 70% Formic Acid (FA) solubilized Aβ40 and 42 (25), cerebral amyloid angiopathy (CAA), apolipoprotein E (APOE) genotype, gender, and age are reported. AD, Alzheimer’s disease; PA, Pathological aging; NDC, non-demented controls; OA, optic atrophy; tr, transynaptic degeneration; GBA, gross brain atrophy; x, below detection levels; ND, not determined, f, female; m, male. Transgenic mice The transgenic mice used in this study have been described previously and are summarized in Table 2. The PrP.APPsi mice express mouse APP-695 cDNA with a humanized Aβ sequence and FAD mutations (APPswe/ind) using the MoPrP.Xho vector, which was co-injected with a vector to express eGFP in skin (first described in (37)). The iTau-P301L mice express human 0N4R tau-P301L using a tet-regulated vector that includes a minimal CMV promoter (38). Tau levels were assessed by Western blotting. PBS lysates of 2.5-6 month old P301L mice were heated at 95°C for five minutes in the presence of denaturing SDS sample buffer, were separated on a 4-12% Bis-Tris gel (Bio-Rad, Hercules, CA) in 3-(N-morpholino)propanesulfonic acid (MOPS) running buffer (Bio-Rad, Hercules, CA) and transferred onto PVDF membrane. The membrane was blocked in casein blocking buffer and incubated overnight at 4°C with primary antibodies to tau, Tau-13 (anti-human tau2-18; Covance) and 3026 (rabbit polyclonal antibody raised against full length 0N3R human tau and also reacts with 0N4R human tau (39, 40)), and anti-β-actin (Sigma-Aldrich, St. Louis, MO) before incubation with secondary antibody, fluorophore conjugated AlexaFluor 680 anti-mouse IgG (Thermo Fisher Scientific, Waltham, MA). Protein bands were detected and quantified using the multiplex Odyssey Infrared Imaging system (Bio-Rad, Hercules, CA). Table 2. Mouse strains used in this study.
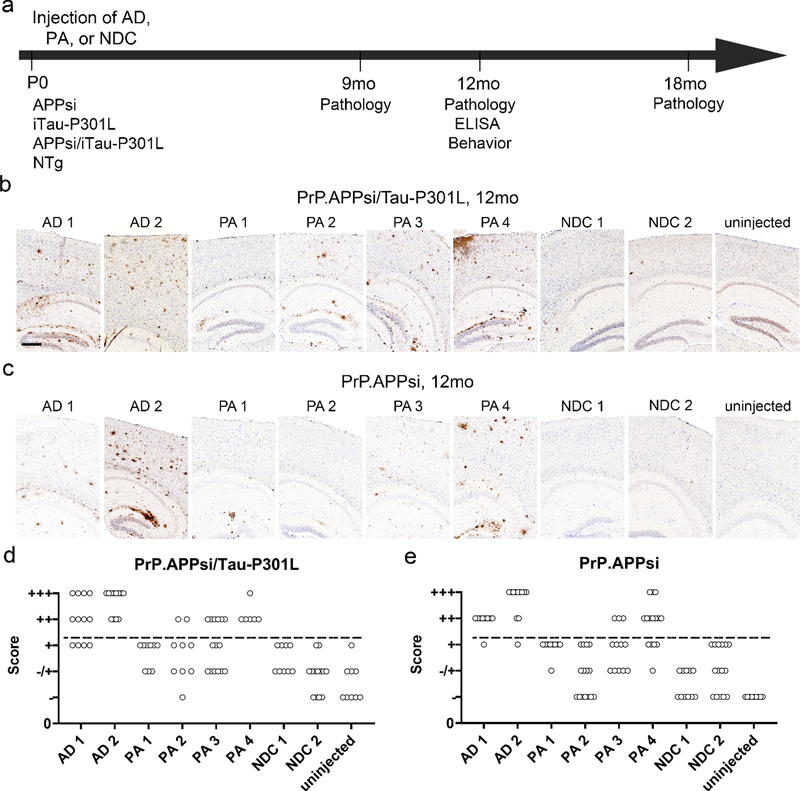
CAA, Cerebral amyloid angiopathy; NA, not applicable. The M20 mice express wild type αSyn under the control of the prion promoter and do not develop αSyn pathology (41). PrP.HuAβ/PS1 mice express human APPswe and human PS1dE9. PrP.MoAβ/PS1 mice express mouse APPswe mutation and human PS1dE9, and tet.MoAβ mice expresses inducible mouse APPswe (37). These mice were bred in-house. PrP.APPsi and M20 were maintained on hybrid C57BL6/C3H backgrounds, following a breeding scheme in which transgene-positive males were bred to nontransgenic F1 B6/C3 female mice. The iTau-P301L mice we used had been backcrossed to the FVB/NCr mouse strain for more than 10 generations. APPsi/Tau-P301L and APPsi/αSyn mice were generated by intercrossing mice that where hemizygous for the respective transgenes. PrP.APPsi mice were genotyped by visualizing GFP expression, which is possible because these mice were co-injected with a transgene that expresses GFP in the skin integrated next to APP transgene. PrP.APPsi/iTau-P301L mice were genotyped for APP by illumination and visualization with special filter goggles (BLS Ltd, Budapest, Hungary) and for tau by PCR of tail DNA. iTau-P301L and M20 mice were genotyped by PCR of tail DNA. PrP.APPsi mice destined for behavioral studies were backcrossed one generation onto C57BL6. All animals were housed 1-5 per cage with unlimited access to food and water with a 14-hour light and 10-hour dark cycle. All experiments involving mice were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC) and conducted in accordance with NIH guidelines. Human brain lysates Human and mouse brain lysates were prepared as previously described (42). Briefly, frozen tissue was homogenized at 10% (w/v) in sterile PBS, subjected to vortex and sonication (3 x 5 sec) and then centrifuged 3000 x g for 5 min at 4°C. Lysates were immediately aliquoted and stored at -80°C until needed. As previously described (25), the Aβ levels and solubility in the lysates was assessed by Western blotting. Briefly, lysates were heated at 50°C for three minutes in the presence of denaturing sample buffer, were separated on a 4-12% Bis-Tris gel (Bio-Rad, Hercules, CA) in 1X2-(N-morpholino)ethanesulfonic acid (MES) running buffer (Bio-Rad, Hercules, CA) and transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA). The membrane was boiled in PBS for 5 min, blocked in Starting Block (Thermo Fisher Scientific [formerly Thermo Scientific], Waltham, MA) and incubated overnight at 4°C with primary antibody Ab5 (human Aβ1-16 specific; T.E. Golde) before incubation with the secondary antibody, fluorophore conjugated Alexa Fluor 680 anti-mouse IgG (Thermo Fisher Scientific, Waltham, MA). Protein bands were detected using the multiplex Odyssey Infrared Imaging system (Bio-Rad, Hercules, CA). Brain seeding with neonatal cerebral ventricle injections Injections of human brain lysates and lysates from a 27mo PrP.APPsi mouse, a 25mo PrP.HuAβ/PS1 mouse, a 24mo Tet.MoAβ mouse, and a 24mo PrP.MoAβ/PS1 mouse were performed as described previously with recombinant adeno-associated virus (43). Briefly, P0 pups were cryoanesthetized and 2 uL of human or mouse brain lysates were bilaterally injected into the cerebral ventricle using a 10 uL Hamilton syringe with a 30-inch needle (Hamilton Company, Reno, NV). Pups were placed on a heating pad for recovery and returned to their home cage. Cohorts of mice were euthanized at 6, 9, 12, and 18 months for analysis. Both male and female mice were analyzed (see Tables S1 and S2 for description of cohorts). Mouse brain tissue collection Mice were anesthetized with isoflurane and perfused transcardially with 20 mL of cold PBS. The brains were cut sagittally through the midline, and one hemibrain was drop-fixed in 4% paraformaldehyde in PBS (pH 7.5) for ~48 hrs at 4°C followed by processing and paraffin embedding. The other hemibrain was snap frozen with isopentane on dry-ice and then stored at -80⁰C until it was thawed and homogenized in preparation for ELISA measurements of Aβ peptide levels. Histology and immunochemistry Paraffin sections (5 μm) were used for all the histology and immunochemistry studies. Campbell-Switzer silver (44) and Thio-S (37) tissue staining methods were performed as previously described. Immunochemistry followed standard protocols described previously (37). To assess amyloid pathology, embedded sections were immunohistochemically stained with a biotinylated pan-Aβ antibody Ab5 (1:500; T.E.G.) and developed using Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) followed with 3,3’-diaminobenzidine (DAB) substrate (Vector Laboratories, Burlingame, CA). To evaluate tau pathology, embedded sections were immunohistochemically stained with 7F2 (1:10,000; Ben Giasson (39)), CP27 (1:1000; Peter Davies), AT8 (1:1000; ThermoFisher Scientific), and MC1 (1:1000; Peter Davies). To assess αSyn pathology, sections were stained with 9C10 (1:1000; Ben Giasson) (31). After development by 3,3’-diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA) substrate and counterstaining with hematoxylin, the slides were coverslipped and images were taken using an Olympus BX60 microscope or scanned by Aperio® XT System (Leica Biosystems, Buffalo Gove, IL, USA) or Zeiss microscope (Carl Zeiss, Oberkochen, Germany) and analyzed using the Zen 2.6 program (Carl Zeiss, Oberkochen, Germany). Blinded observers then reviewed the images and scored the burden of Aβ pathology based on the following criteria: “+++” = heavy Aβ burden with too many deposits to count. “++” = abundant pathology with >30 deposits per section. “+” = consistent pathology with > 5 deposits per section. “+/-” = inconsistent pathology with <3 deposits per section. Brain amyloid extraction Human brain samples were originally analyzed by Moore (25). Briefly, frozen pre-frontal cortex tissue was cryo-pulverized in liquid nitrogen and then sequentially extracted with Tris-buffered saline (TBS), radioimmunoprecipitation buffer (RIPA), 2% sodium dodecyl sulfate (SDS), and 70% formic acid (FA) containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA). After harvesting the mouse brain, the left hemisphere was flash-frozen in isopentane. The frozen cortex was sequentially extracted with protease inhibitor cocktail (Roche) containing Tris-buffered saline, RIPA buffer, 2% SDS, and 70% formic acid (FA) as described previously at a concentration of 150 mg/ml (25). ELISA Aβ levels from the 2% SDS and 70% FA extracted samples were quantified by sandwich ELISA as described previously (25). Total Aβ was captured with mAb Ab9 and detected by HRP-conjugated mAb 4G8 (human Aβ17-24; Covance, Princeton, NJ, USA); Aβ1-40 was captured with monoclonal antibody (mAb) Ab9 (human Aβ1-16; T.E. Golde) and detected by horseradish peroxidase (HRP)-conjugated mAb 13.1.1 (human Aβ35-40 specific; T.E. Golde); Aβ1-42 was captured with mAb 2.1.3 (human Aβ35-42 specific; T.E. Golde) and detected by HRP-conjugated mAb Ab9. ELISA results were analyzed using SoftMax Pro software (Molecular Devices). Contextual fear conditioning Transgenic PrP.APPsi and nontransgenic littermates, seeded with AD cases 1 and 2, and NDC case 7, were aged to 12 months and subjected to contextual fear conditioning as previously described (45, 46). Briefly, in the contextual fear conditioning test, mice learn the association between the training chamber, which represents an initially neutral conditional stimulus (CS) and an aversive, brief electric foot-shock, unconditional stimulus (US), which takes place in the training context. We have previously established that 0.45 mA electric current elicits robust avoidance response and results in strong freezing response after 2 CS-US pairings (45). During the training session, mice were allowed to explore the training chamber for 120 sec, with a 2 sec foot shock immediately following a 30 sec tone (80 dB) (day 1; see Fig. 4A). The mice recover for 60 sec and then receive another 2 sec foot shock following a 30 sec tone. The final post-US period is 60 sec. For the context test session, on day 3, mice were exposed to the same training context and the freezing presentation during the 300 sec was recorded by an image analysis system (FreezeFrame, Actimetrics). On day 4, mice experienced the tone session, and were placed in a modified chamber (altered inserts and smell), during the first 180 sec the mice were allowed to explore the new environment. During the second 180 sec period the tone was delivered and the percent freezing was recorded. No shock was applied during context or tone session on days 3 and 4. Results Characterization of AD/LBD, AD, PA, and NDC cases In this study, we selected a subset of AD, PA, and NDC donors that were previously analyzed (25) for examination of seeding activity in APP, APP/tau, and tau transgenic mice (Table 1, AD 1 and 2, PA 1, 2, 3, 4, and NDC 1 and 2). All 8 of these donors exhibited some degree of Lewy body pathology (LBD). Two of the donors selected for the study were sub-categorized with cerebral amyloid angiopathy (CAA); PA 3 had a high abundance of vascular amyloid with a CAA score of 2+-3+ while AD 1 had moderate levels of vascular Aβ deposition with a score of 1+ (Table 1). As expected, the two AD brains had significant accumulation of tau, indicated by Braak staging (5.5 and 6) (Table 1). The PA and NDC brains had lower abundance of tau than the AD brains, but were similar to each other (2, 2.5, 3, 2, and 2, 3, respectively) (Table 1). In this set of cases, all also exhibited incidental, diffuse, αSyn pathology. To these 8, we added 6 additional donors that included two AD (no LBD), two with Lewy-body variant AD (AD/LBD), and 2 additional controls that were free of all types of pathology (Table 1, AD 3 and 4, AD/LBD 1 and 2, and NDC 3 and 4). These brains were used in seeding APP, APP/α-synuclein (αSyn), and αSyn mice so that we would be able to compare AD and NDC that lacked αSyn pathology to AD/LBD brains with high levels of αSyn pathology. To characterize the pathology in the AD and PA brains used to seed the APP/tau, APP, and tau mice, sections were stained with a pan-Aβ antibody (Fig. S1a), Thioflavin S (Thio-S) (Fig. S1b), and with a phospho-specific tau antibody (Fig. S1c). We observed widespread Aβ deposition of both diffuse and compact amyloid in both AD and PA (Fig. S1a, b), with a subset showing striking CAA. PA 3 had numerous, Thio-S positive, vascular amyloid deposits, consistent with the assessment of CAA as 2+-3+ (Fig. S1a, b). Similarly, we observed Thio-S positive amyloid staining surrounding several vessels within AD 1, consistent with a CAA score of 1+ (Fig. S1a, b). AD 2 contained several cored Thio-S positive deposits while the compact deposits in PA 1, 2, and 4 had little to no Thio-S staining. As expected, AD 1 and 2 contained both substantial phosphorylated tau in form of dystrophic neurites, neuropil threads and neuronal inclusions while all four of the PA were negative for tau deposits (Fig. S1c). NDC 1 and 2 were negative for Aβ and tau deposits by both immunostaining and Thio-S staining. Cerebral injection of brain lysates into newborn APP/tau and APP mice results in widespread, robust amyloid deposition To gain a better understanding of the type of Aβ pathology that unseeded PrP.APPsi mice produce, we harvested breeder mice at various ages to assess phenotypic variation (Fig. S2a and b) (37). At 12 months of age, when deposition begins to occur in the brains of PrP.APPsi mice, any given section through the cortex and hippocampus may exhibit 1 or 2 diffuse tufted deposits and/or cored deposits (score +/-). At this age, Aβ deposition also begins to appear in the meninges surrounding the cerebellum (Fig. S2a). Approximately 50% of PrP.APPsi mice at 11-13 months of age exhibit no Aβ pathology or show only meningeal deposition in the cerebellum (Fig. S2a). To concurrently examine whether tau pathology could be augmented by seeding from these lysates, the PrP.APPsi mice were crossed to a line of mice that express human tau P301L (iTau-P301L) (Table 2) (38, 47). The iTau-P301L mice express mutant human tau at levels 2-3 fold higher than nontransgenic mice (Fig. S3). To compare the relative ability of homogenates prepared from AD and PA cases to seed Aβ deposition in these mice, lysates from the pre-frontal cortex of the AD, PA, and NDC were injected into the cerebral ventricles of newborn mice at P0 (31, 42, 43). We confirmed that the lysates contained Aβ by immunoblotting and ELISA (Fig. S1d and Table 1). Aβ was detectable by immunoblot in each of the lysates used for injection (Fig. S1d), and enzyme-linked immunosorbent assay (ELISA) measurements confirmed high levels of SDS-soluble and formic acid (FA) soluble Aβ42 in each brain lysate (Table 1). We hypothesized that injection of these lysates in P0 mice would result in widespread dispersion of the Aβ or tau seeds, and potentially offer the best chance to extensively alter the type of pathology that would be induced by seeding. To assess amyloid pathology, hemibrains of seeded mice were stained with a pan-Aβ antibody and Thio-S. In mice injected with either AD or PA lysates, we observed significant induction of Aβ deposition, comprised primarily of diffuse deposits throughout the cortex and hippocampus, with some vascular deposition in the pia surrounding the cortex and within the hippocampal fissure (Fig. 1b, c). Mice injected with homogenates from NDC 1 and 2, and mice that were not injected, had little Aβ pathology by 12 months of age (Fig. 1b, c). Examples of cases with the most severe pathology are shown in Figure 1, while images of animals with the least pathology are shown in Figure S4. In all cases, however, the induced Aβ pathology exhibited a diffuse, Thio-S negative morphology (Fig. S5). The levels of induced Aβ pathology in mice that co-expressed APPsi and Tau-P301L were similar to that of mice that expressed only APPsi (Fig. 1b versus c, S4a versus b). Mice that were transgenic for only Tau-P301L, or were non-transgenic, showed no evidence of Aβ deposition (data not shown). To compare the data across all of the mice examined, Aβ pathology was qualitatively scored by three independent observers (Fig. 1d, e). At 12 months, only PA 4 was scored as having a level of Aβ pathology that approached mice injected with the AD cases; both of which exhibited widespread, diffuse pathology (Fig. 1b, c). Aβ pathology in mice seeded by PA 1, 2, and 3 was scored as less abundant than in mice seeded by the AD cases (Fig. 1d, e). We confirmed the high seeding activity of AD 1 and 2, and PA 4 by assessing Aβ pathology at 9 months post-injection. Although the severity of pathology varied, multiple animals injected with each of these lysates exhibited Aβ deposition (Fig. S6a). Notably, dilution of the brain lysates by 10-fold prior to injection greatly diminished seeding activity (Fig. S6b). To further examine the relative seeding activity of PA lysates, we injected newborn PrP.APPsi mice with each of the 4 PA lysates and NDC 1, and analyzed pathology at 18 months post-injection. Although the mice injected with the PA lysates appeared to have more severe Aβ deposition, we also observed some significant Aβ pathology in animals injected with the NDC lysate (Fig. S7). Whether this outcome was due to some small amount of Aβ seed in the NDC 1 will require further study. Collectively, these studies demonstrated that AD and PA brains contain misfolded forms of Aβ that preferentially seed diffuse Aβ pathology.
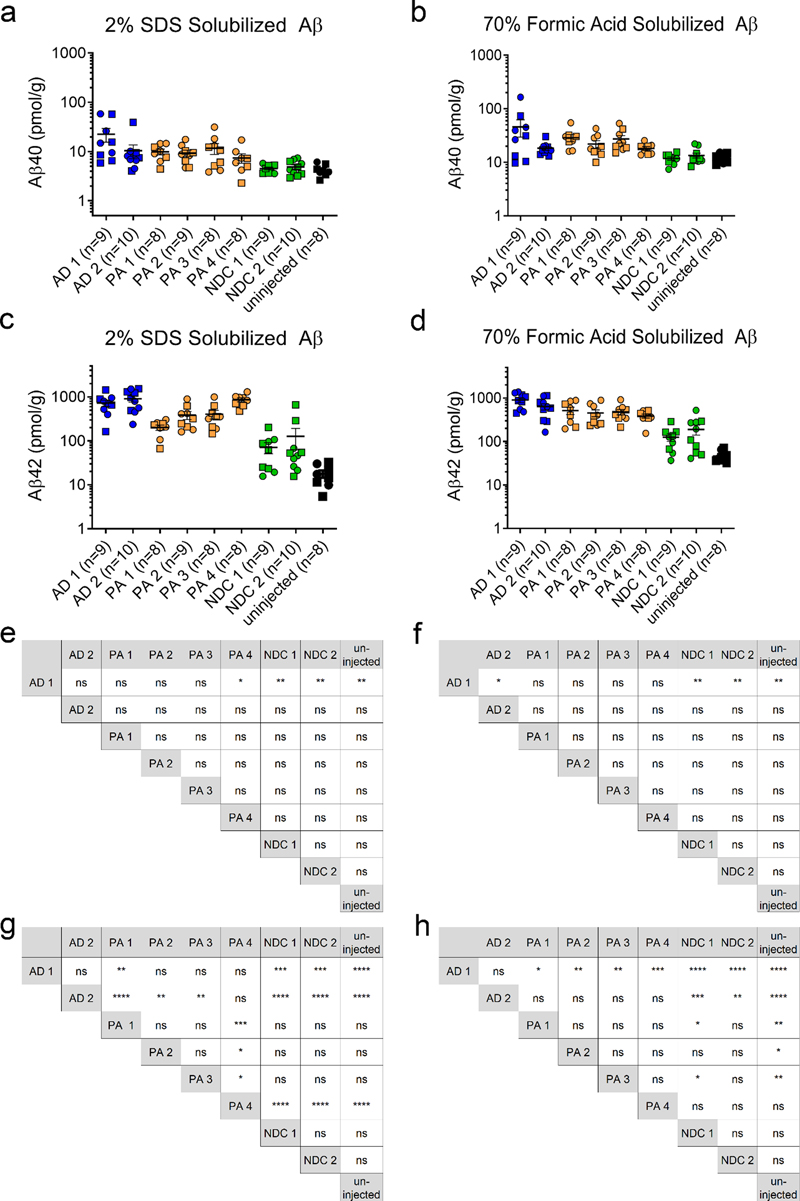
Fig. 1 Cerebral injection of brain lysate at P0 induces widespread, robust amyloid deposition. Overall schematic of experiments (a) Images of PrP.APPsi/Tau-P301L (b) and PrP.APPsi (c) mice injected with AD, PA, or NDC lysate at P0. Brain sections (hemibrain) stained with biotinylated anti-Aβ mAb Ab5 (anti-Aβ 1-16) and counterstained with hematoxylin. Cases with the most abundant amyloid pathology are shown. Scale bar: 100 μm. Qualitative analysis of amyloid deposition in seeded PrP.APPsi/Tau-P301L (d) and PrP.APPsi (e) mice. Three observers independently scored representative slides from each seeded mouse. The amyloid pathology in each mouse was categorized on a scale from (-) as no pathology to (+++) as abundant pathology. Each point represents a rating score by an observer (3 observations per animal). The number of seeded mice for each scale of amyloid burden by the three observers is reported. A subset of mice injected with NDC lysate were rated as + based on 3-5 plaques per section, and thus we set this rating as baseline (marked by a dash line). Only the two AD cases and PA 4 consistently scored above +. The number of PrP.APPsi/Tau-P301L or PrP.APPsi mice injected with each lysate, respectively, were as follows: AD 1: n=4, n=3; AD 2: n=4, n=3; PA 1: n=3, n=3; PA 2: n=3, n=5; PA 3: n=5, n=4; PA 4: n=2, n=5; NDC 1: n=3, n=3; NDC 2: n=5, n=5; uninjected: n=3, n=3. Two of the donor cases were identified as having CAA pathology, AD 1 and PA 3, and were scored 1+ and 2+-3+, respectively (Table 1). To determine if CAA pathology was seeded in the recipient mice, we stained sections with Thio-S and searched for evidence of vascular amyloid. In mice seeded with PA 3, we observed Thio-S positive, vascular pathology in 6 of the 7 recipient mice; however, the incidence of this pathology was limited (Fig. S8). Although AD 1 also contained CAA pathology, we did not observe Thio-S positive, vascular pathology in mice seeded by this homogenate. These findings suggest that it may be possible to selectively increase CAA using seeding, but enhancing such pathology may require purification of cerebral vessels before preparation of the seeds. We biochemically confirmed our histological data by analyzing Aβ levels from sequentially extracted brain lysates using C-terminal specific antibodies in sandwich ELISAs to measure Aβ40 and Aβ42 specifically. In our Aβ ELISAs the detection limit was approximately 0.04 pmol/g. In the brains of all seeded mice, the levels of Aβ40, both SDS-soluble and FA, were 10-100-fold lower than that of Aβ42 (Fig. 2a, b). Consistent with the presence of abundant diffuse Aβ pathology, we detected elevated levels of Aβ42 in SDS-soluble fractions from mice seeded with AD 1 and 2, and PA 4 (Fig. 2c). Somewhat surprisingly, the levels of Aβ42 in the FA-soluble fractions were similar to that of the SDS fraction despite the absence of cored Aβ deposits (Fig. 2c and d). Notably, there was considerable variation in the levels of Aβ in these seeded animals, which rendered relatively few indications of statistical differences (Fig. 2e-h). For Aβ40 measures, the only instance in which the seeded mice had levels that were higher than uninjected controls, or mice injected with NDC lysate, were mice seeded by AD 1 (Fig. 2e, f). For measurements of SDS-soluble Aβ42, the brains of mice seeded with AD 1, 2, and PA 4 were the only examples in which the levels were statistically higher than the levels in mice seeded with NDC 1 and 2, or uninjected mice (Fig. 2g). In the FA-soluble fractions, only the AD lysates possessed higher levels of Aβ42 than the brains of mice seeded with the two NDC lysates or uninjected mice (Fig. 2h). Collectively, these findings demonstrated that both AD and PA brains have the potential to seed diffuse Aβ pathology, with the AD brains appearing to be slightly more potent.
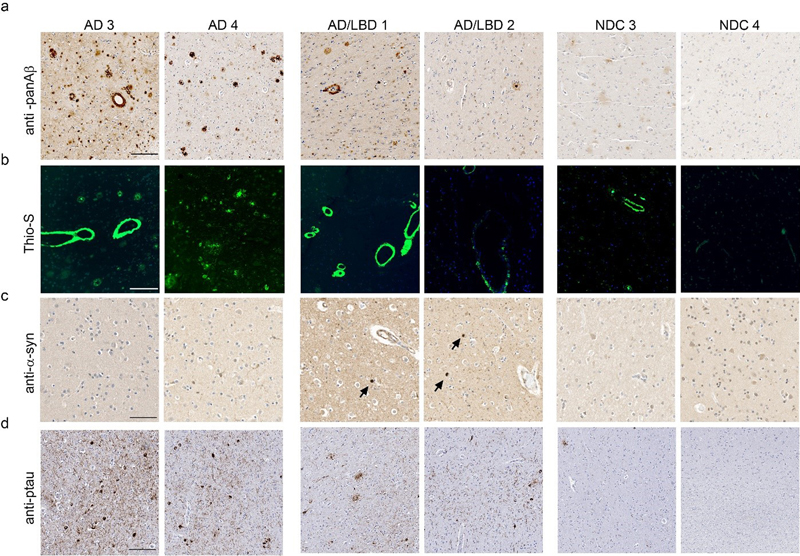
Fig. 2 Biochemical analysis of sequentially extracted Aβ42 and Aβ40 levels by end-specific sandwich ELISA. PrP.APPsi/Tau-P301L and PrP.APPsi mice were seeded by AD, PA, or NDC lysate at P0 and aged 12 months. (a, e) 2% SDS-extracted Aβ40, (b, f) 70% formic acid Aβ40, (c, g) 2% SDS-extracted Aβ42, and (d, h) 70% formic acid Aβ42. Data plotted as scatter dot plot of PrP.APPsi/Tau-P301L (square) and PrP.APPsi (circle) ± standard error of the mean. The number of PrP.APPsi/Tau-P301L or PrP.APPsi mice injected with each lysate, respectively, were as follows: AD 1: n=5, n=4; AD 2: n=6, n=5; PA 1: n=5, n=4; PA 2: n=4, n=5; PA 3: n=5, n=4; PA 4: n=3, n=5; NDC 1: n=5, n=4; NDC 2: n=5, n=5; uninjected: n=5, n=3. Aβ42 and Aβ40 levels were quantified with corresponding one-way ANOVA with multiple comparisons test (ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001). To determine whether the injection of these lysates into the brains of newborn PrP.APPsi/iTau-P301L mice also induced tau pathology, we stained the brains of the seeded animals with antibodies specific for phosphorylated tau (AT8) and misfolded tau (MC1). Immunostaining with the antibody CP27, which is specific for human tau, confirmed the presence of human P301L tau in the bigenic mice, but we observed no obvious reactivity with AT8 or MC1 antibodies (Fig. S9). To determine if the induction of amyloid would secondarily induce αSyn pathology, we examined the seeding activity of individuals with AD and LBD pathology. To characterize the pathology of these AD, AD/LBD, and NDC brains, we stained sections with a pan-Aβ antibody (Fig. 3a), Thio-S (Fig. 3b), αSyn antibody (Fig. 3c), and a phospho-specific tau antibody (Fig. 3d). Both AD 3 and 4 contained numerous amyloid deposits that were compact and Thio-S positive (Fig. 3a, b), had widespread tau positive inclusions (Fig. 3d), and lacked αSyn pathology (Fig. 3c). AD 3 showed striking Thio-S positive amyloid staining surrounding vessels, consistent with a CAA score of 3+ (Fig. 3b, Table 1). We also observed CAA staining with AD/LBD 1 (Fig. 3b). The AD/LBD cases showed modest Aβ pathology with some αSyn pathology and sparse tau deposition (Fig. 3a, c, d). NDC 3 contained some amyloid pathology, but both lacked αSyn and tau pathology (Fig. 3).
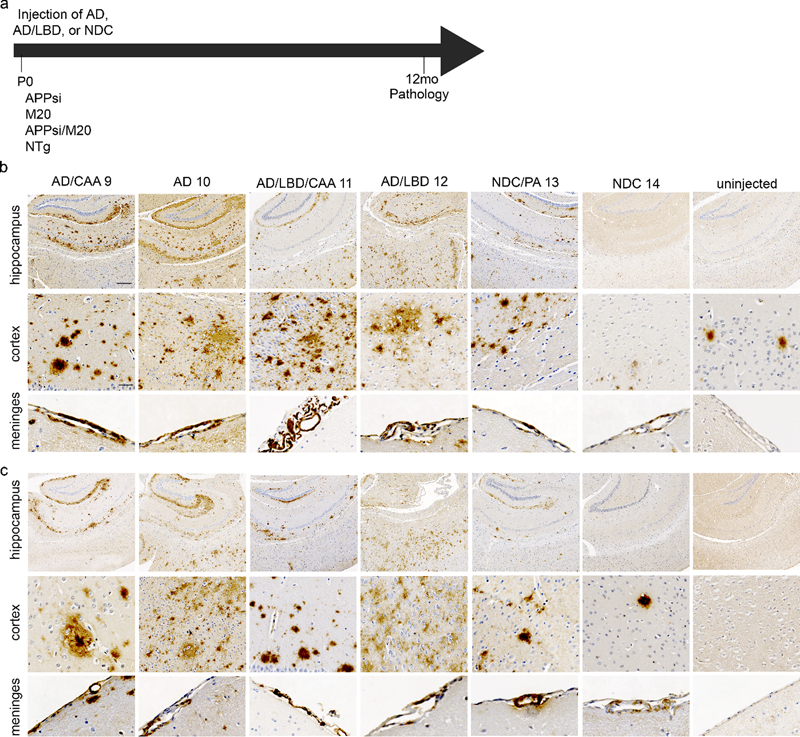
Fig. 3 Characterization of AD, AD/LBD, and control cases. Representative image from the cortex of 2 AD cases, 2 AD/LBD cases, and 2 NDC cases stained with (a) biotinylated anti-Aβ mAb 33.1.1 (anti-Aβ 1-16). Scale bar: 100 μm, (b) Thio-S. Scale bar: 50 μm, (c) anti-αSyn mAb 9C10 (anti αSyn 2-21). Arrowheads depict Lewy bodies. Scale bar: 100 μm, and anti-ptau mAb 7F2 (pThr205). Scale bar: 150 μm. PrP.APPsi/M20 and PrP.APPsi mice injected with AD brains developed widespread, robust Aβ pathology by 12 months post-injection (Fig. 4a and b). Aβ deposition was primarily diffuse and Thio-S negative (Fig. S10). There was no difference in Aβ seeding capacity between PrP.APPsi/M20 and PrP.APPsi mice. Interestingly, AD/LBD 1 and 2 also promoted deposition of Aβ in mice expressing APPsi with and without αSyn (Fig. 4), indicating that the AD/LBD brains contained considerable levels of Aβ seeds despite a much lower burden of Aβ pathology (Fig. 3). None of the injected animals developed appreciable αSyn pathology (data not shown). Although two of the donor cases contained significant CAA pathology, CAA was not seeded in the recipient mice (Fig. S10).
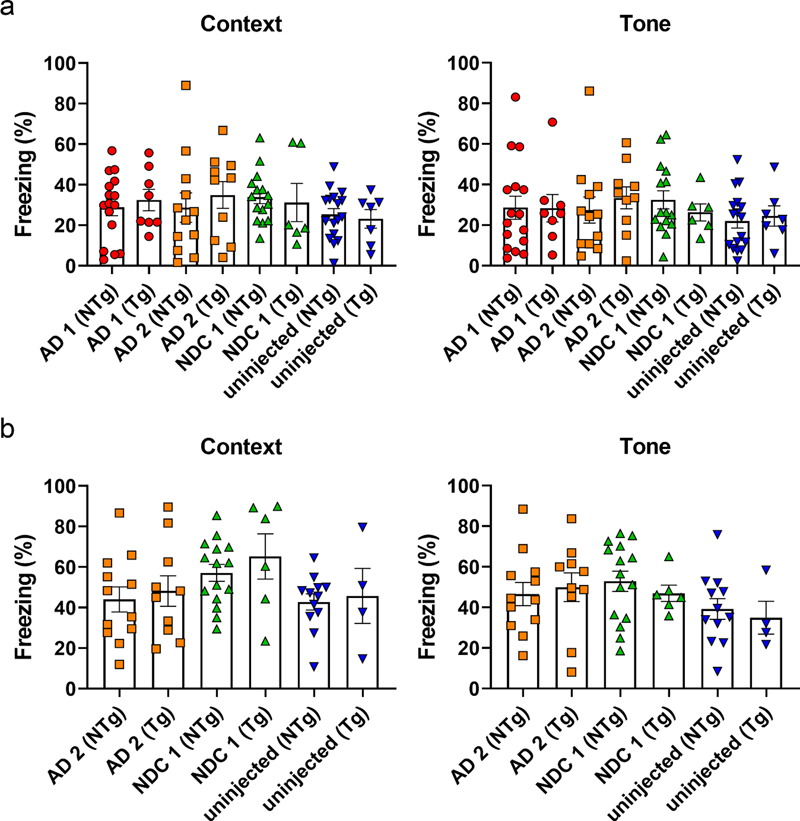
Fig. 4 Cerebral injection of AD and AD/LBD brain lysates results in widespread amyloid deposition and CAA. Overall schematic of the experiment (a). Newborn P0 mice were injected with AD, AD/LBD, and NDC brain lysates and aged 12 months. Representative brain sections (hippocampus, cortex, and meninges) of (b) PrP.APPsi/Line M20 and (c) PrP.APPsi mice stained with biotinylated anti-Aβ mAb Ab5 (anti-Aβ 1-16) and counterstained with hematoxylin. n=4-8. Scale bar: hippocampus: 250 μm, cortex and meninges: 50 μm. Seeding diffuse Aβ deposition does not impair performance in a fear-conditioning cognitive task To assess whether the diffuse Aβ pathology seeded by AD brain produces cognitive deficits, an additional cohort of PrP.APPsi mice were seeded with AD 1 and 2, or NDC 1, and then were aged 12 months before behavioral testing in a fear-conditioning paradigm. Control groups were mice that were not injected with brain lysate and nontransgenic littermate mice that were either injected with the same lysates or uninjected. The performance of the animals in the contextual fear memory test was conducted twice. In the first round of testing, the performance of the seeded mice was not statistically different from that of the control uninjected mice (Fig. 5a). Overall, in this first round of testing, the percentage of time animals that exhibited freezing behavior was relatively low. In the second round of testing, we expected to reinforce memory of the adverse cue (context or tone) and we observed that re-tested mice showed increased levels of freezing behavior (Fig. 5b); however, there was still no statistically significant difference in the performance of the seeded mice relative to any control. These findings indicate that the diffuse Aβ pathology seeded by these homogenates of human AD brain does not produce a meaningful impairment in the systems involved in fear-conditioning memory tasks.
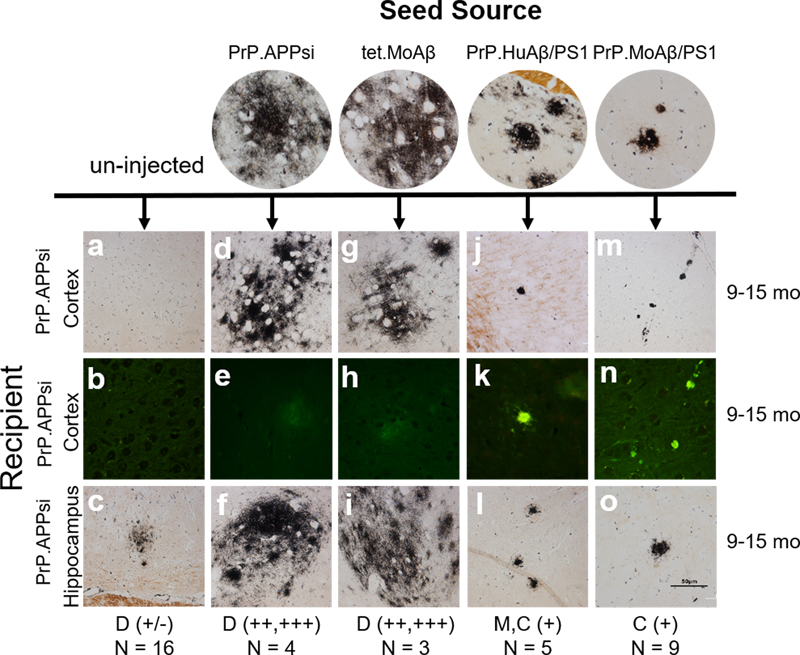
Fig. 5 Diffuse amyloid seeding does not cause cognitive impairment in fear-conditioning. PrP.APPsi mice seeded by AD and NDC lysates, were aged 12 months and subjected to contextual fear conditioning. Mean percentage freezing ± standard error of the mean exhibited by PrP.APPsi and nontransgenic (NTg) littermates injected with AD cases 1 or 2 and NDC case 7. Uninjected mice were the control group. (a) Context and tone paradigm. (b) A subset of mice were re-tested the following week, context and tone test. n = 6-8/Tg, n = 12-16/NTg mice per group. Assessment of Aβ seeding efficiency in APPsi mice To examine the relative seeding efficiency of different types of Aβ pathology in PrP.APPsi mice, we used an approach in which homogenates from the brains of 4 different lines of aged APP and APP/PS1 mice were used to seed accelerated Aβ deposition (described in Table 2). At these advanced ages, each of the mice used to produce inoculum had substantial Aβ pathology (Fig. S11). Following the strategy used above, we injected the homogenates containing Aβ seeds into newborn mice on neonatal day 0, which were then aged to 9, 12, or 15 months before euthanasia and neuropathological analysis by Campbell Switzer silver stain and Thio-S staining. The type of pathology noted and the relative abundance score are noted at the bottom of each column of images in Figure 6. In all of the PrP.APPsi mice injected with brain homogenate seeds from mice that primarily exhibited cored plaques, we observed an obvious shift in the type of Aβ deposits in recipient mice to match the pathology found in the donor mice used for seeding (Fig. 6). Self-seeding of newborn PrP.APPsi mice with brain homogenates from aged PrP.APPsi mice produced diffuse deposits (Fig. 6), which was the predominant form of Aβ pathology in the PrP.APPsi animal used to generate the seeding homogenate (Fig. S11). Similarly, the predominant pathology in the ~25 month old Tet.MoAβ animal used to prepare seeding homogenate was diffuse Aβ deposits (Fig. S11)(48), and brain homogenates from this animal efficiently seeded the deposition of human Aβ in the PrP.APPsi mice to produce diffuse deposits (Fig. 6). PrP.APPsi mice seeded with homogenates from donors that had primarily cored deposits had lower Aβ burden scores, but importantly we observed a shift in the neuropathologic features of the recipient mice to a much higher incidence of cored deposits (Fig. 6). Interestingly, the morphology of deposits in the seeded PrP.APPsi mice was not quite identical to the source PrP.HuAβ/PS1 mice in that the seeded deposits were smaller and appeared to be more compact than the donor line. These small dense deposits were Thio-S positive (Fig. 6). Similar small dense core deposits that were Thio-S positive were observed in the cortex of PrP.APPsi mice injected with brain homogenates from the PrP.MoAβ/PS1 mice (Fig. 6). Collectively, these findings demonstrate the relative ease with which diffuse Aβ pathology can be induced in PrP.APPsi mice.
Fig. 6 Comparison of seeding activity between transgenic mice that exhibit diffuse versus cored Aβ pathology. We selected brains from four sources that exhibit either diffuse or cored neuritic Aβ deposits (see Fig. S11). Newborn PrP.APPsi mice were injected with homogenate from each of the four sources identified at the top of the figure. Compared to uninjected mice, PrP.APPsi mice injected with brain homogenates from any source showed an induction of Aβ pathology. Inoculum from aged PrP.APPsi and tet.MoAβ mice, which primarily exhibit diffuse Aβ pathology (37, 48), robustly seeded diffuse pathology in the injected PrP.APPsi mice (severity of pathology and number of animals indicated at the bottom of the figure). Inoculum from aged PrP.HuAβ/PS1 (a.k.a. APPswe/PS1dE9) and PrP.MoAβ/PS1 mice, which primarily exhibit cored neuritic deposits (37, 48), induced the deposition of a limited number of cored Aβ deposits in injected PrP.APPsi mice with little or no diffuse deposits. D = diffuse Aβ pathology. M = mixed diffuse and cored deposits. C = cored, neuritic deposits. Discussion We investigated whether injection of neonatal APP, APP/tau, or APP/αSyn mice with AD or AD/LBD brain homogenates could be used as a paradigm to generate mice that model the mixed pathology associated with each disease. Despite the widespread induction of diffuse Aβ deposition by injection of four different AD brain lysates, and the presence of tau seeds in these lysates, tau pathology was not induced. For comparison, we also injected PA brain lysates, finding a similar induction of diffuse Aβ deposition without induction of tau pathology. Similarly, injection of AD/LBD brain lysates induced diffuse Aβ deposition, but αSyn pathology was not induced. In all cases, the induced Aβ pathology exhibited a diffuse, Thio-S negative, morphology. PrP.APPsi mice injected with AD brain lysates that develop diffuse Aβ pathology showed no significant impairment in a fear-conditioning cognitive task. Our findings suggest that seeding the brains of neonatal transgenic mice with AD, AD/LBD, and PA brain homogenates can efficiently induce diffuse Aβ deposition but neither AD nor AD/LBD brains seeded concurrent tau or αSyn pathology. The most common route of administering Aβ seeding preparations is by stereotaxic injection into the hippocampus of adult APP transgenic mice (reviewed in (13)). In host mice that develop amyloid pathology at relatively young ages, the induced pathology created by seeding may be localized to the site of injection and overlying cortex (13); whereas, in models that do not develop deposits until late in life, the induced pathology may be more wide-spread (reviewed in (13)). The PrP.APPsi host we used in the present study develops pathology on its own between 12-14 months of age. In testing the approach of injecting Aβ seeds into newborn mice from this model, we aspired to attain a widespread distribution of seeding material and induce Aβ, tau, or αSyn pathology throughout the brain. This approach replicates previous studies where P0 injection of adeno-associated virus resulted in widespread distribution in the brain (49). Injection of newborn hamsters with scrapie prions has been shown to accelerate onset of prion disease (50). Additionally, we have observed that intracerebral injection of newborn A53T αSyn mice with brain lysates from multiple system atrophy donors induces αSyn pathology and motor impairment (31), and that intraspinal injection of newborn mice expressing mutant superoxide dismutase (SOD1) can accelerate the onset of paralysis and pathology (51, 52). Thus, in performing newborn injections our goal was to initiate pathological cascades as early as possible in order to determine whether there may be distinct synergies between Aβ and tau, or Aβ and αSyn, which can be elaborated by seeding. We recognize that there are a large number of potential mouse models that could have been used for these studies. The APP mouse model we used for these studies was selected because PrP.APPsi mice do not inherently develop Aβ pathology until 12-14 months of age and because this model can exhibit a full spectrum of Aβ pathology including cored-neuritic, diffuse, and vascular deposition (see Supplemental Fig. S2). The tau model we chose to use expresses human P301L tau at relatively low levels and does not develop tau pathology on its own. When paired with mice that express the tetracycline transactivator in the rTg4510 model these develop a robust tau pathology (38). In paradigms in which tau expression in rTg4510 mice is induced early and then suppressed by doxycycline, the low level of “leaky” tau expression in this model is sufficient to sustain neurofibrillary tangle pathology (38). In our paradigm, we asked whether neonatal seeding of this tau model could induce a sustained pathology in the same way that early expression of the transgene at high levels produced sustained pathology. Alternatively, we were interested to determine whether we could detect any synergy between concurrent Aβ pathology and tau seeding. The αSyn model we chose to use expresses human WT αSyn at levels that do not cause pathology (41). WT αSyn (M20) mice can be seeded when injected with high levels of purified αSyn fibrils, but are not easily seeded by human brain homogenates (31). Our goal in choosing the M20 WT αSyn mice was to develop a model of human αSyn pathology and assess whether concurrent Aβ pathology could synergize αSyn seeding. Our method of seed preparation followed commonly used protocols where we injected a soluble fraction; frontal cortex homogenized in PBS (10% w/v) followed by sonication and low speed centrifugation, as previously described (42, 53, 54). By this method, we expected sonication to fragment all types of seeds with a mixture of seeds remaining in suspension after low-speed centrifugation. As we have observed here, the most common outcome of Aβ seeding with similar preparations is induction of diffuse Aβ pathology (reviewed in (13)). In order to efficiently seed mixed pathology, it may be necessary to optimize preparations of each type of seed independently. Interestingly, injection of the supernatant and pellet of fractionated APP23 brain homogenate resulted in morphologically different Aβ deposits, with the supernatant fraction seeding diffuse, Congo Red negative Aβ and the pellet seeding deposits similar to the total lysate, a mixture of diffuse and punctate, Congo Red positive deposits (55). Several studies have shown induction of tau pathology after injection of synthetic tau fibrils, brain extract from mutant P301S tau mouse, or human AD, corticobasal degeneration, and progressive supranuclear palsy brain lysates (56-62). In these studies, the human brain lysates were enriched for tau seeds by sequential fractionation with the addition of sucrose and/or sarkosyl, creating fractions that contain potent tau seeds. Similarly, injection of the fractionated homogenate from brain lysates of individuals with LBD resulted in the induction of αSyn, indicating that with enrichment it is possible to seed αSyn pathology directly from human brain (20, 63, 64). Because one of our goals was to determine whether we could detect synergy between Aβ and tau or αSyn pathology, we chose to avoid enriching for any particular type of seeding activity. The absence of tau or αSyn pathology in our seeded models indicates that the type of Aβ deposition we generated did not synergize to induce, or exacerbate, tau or αSyn pathology. We demonstrated that the Aβ seeding activity of brain lysates from PA cases was similar to AD and AD/LBD cases. These findings agree with a previous study that reported brain homogenates from a human PA case seeded diffuse Aβ deposition in Tg2576 APP mice (65). Although mice seeded by PA brains scored as having less amyloid positivity at 12 months post-injection than mice injected with AD brain lysates, the difference between PA and AD brain seeding activity was less evident by 18 months post-injection. Our study was not powered or designed to determine whether homogenates from AD and PA brains have quantitative differences in seeding activity. All three of the PA cases tested were able to seed amyloid pathology, supporting the hypothesis that the Aβ pathology in PA is similar to AD (25). Interestingly, several of the brain lysates we used were from tissues that exhibited relatively high levels of CAA pathology (see Table 1). PA 3 and AD 3 were both scored as having the highest levels of CAA, with significant CAA in AD/LBD 1 and 2. In mice injected with PA 3, we observed modest seeding of parenchymal CAA pathology in the host mice but in all other mice the only obvious vascular deposition was meningeal. Augmented CAA pathology has been observed in APP23 mice, which develop CAA, by intraperitoneal injection of APP23 lysates (66). CAA pathology was also induced in CRND8 mice by injection of synthetic Aβ42 oligomers, generated in the presence of anionic micelles composed of fatty acids (67). Recent studies have documented early onset cerebrovascular Aβ pathology in individuals receiving dura mater transplants and cadaveric pituitary-derived growth hormone, suggesting that CAA may be transmitted iatrogenically (68, 69). Collectively, these results suggest that CAA could be a distinct conformer of Aβ that may be independently propagated. Not all Aβ conformers seeded as proficiently as vascular Aβ. Tissues from AD and AD/LBD brains that were used to prepare these lysates contained both diffuse and compact Aβ deposits, with significant levels of formic acid extractable Aβ. Yet, even in mice aged to 18 months of age, diffuse Aβ deposits were the dominant form of pathology. These results mirror similar studies in which injection of human brain homogenates resulted in induction of diffuse Aβ pathology, with relatively few Thio-S or congophilic compact deposits (49, 65, 70, 71). The lack of conversion of the diffuse deposits to cored plaques indicates that these types of amyloid deposition are not freely interchangeable, supporting the idea that each type of Aβ pathology arises from different amyloid strains with distinct seeding capabilities. This conclusion is also supported by our studies of Aβ seeding in which transgenic mice were used as the seed source. Newborn PrP.APPsi mice injected with transgenic mouse brain homogenates prepared from mice that exhibit high levels of diffuse Aβ pathology produced a robust induction of diffuse Aβ deposition by 12 months post-injection. By contrast, brain homogenates prepared from mice that predominantly produce cored, Thio-S positive, Aβ deposits induced minimal Aβ deposition by 12 months. These results are consistent with previous studies with APP/PS1 and APP23 mice. Injection of APPPS1 mice that typically develop compact, punctate plaques, with APP23 seeds results in a mixture of diffuse, filamentous Aβ as well as compact plaques (42, 72). Injection of APP23 mice that typically develop mixed pathology, both cored and diffuse plaques, with APPPS1 homogenate results in plaques that are more diffuse than the punctate deposits seen in APPPS1 mice injected with APPPS1 homogenate (42, 72). Thus, although it is possible to seed cored deposits in APP mice (42, 55, 73-75), it appears that diffuse Aβ pathology is more easily seeded. Together these results suggest that Aβ plaques differ in their morphology, seeding ability, and impact on cognitive function. Current amyloid therapies aim to reduce amyloid deposition without much consideration of the type of plaque that is being targeted. Reduction of diffuse amyloid may at first appear to be beneficial, since it appears to be the dominate seeding strain, however, it may not be the most pathological strain. Cognitive behavior studies of the seeded APPsi mice reported here, and of a non-seeded Bri-Aβ42 model that also shows primarily diffuse Aβ pathology (76), failed to associate diffuse amyloid with reduced performance in a fear-conditioning memory task. If diffuse Aβ pathology is less damaging, then therapies that only reduce this pathology may not produce therapeutic benefit in AD. Conclusions In conclusion, we assessed the type of pathology induced when neonatal APP transgenic mice were injected with brain lysates prepared from AD, AD/LBD, and PA brains, finding robust and widespread induction of Aβ pathology. The induced Aβ pathology was diffuse, which has been reported in other seeding studies where adult animals were the recipients of injections (13). Our results are consistent with earlier reports in finding that diffuse Aβ is easily seeded by crude homogenates of human brain. The diffuse Aβ pathology induced in these models was not accompanied by secondary tau or αSyn pathology in bigenic mice co-expressing APP/tau or APP/aSyn. Seeded mice with diffuse Aβ pathology were not impaired in a fear-conditioning memory task. The seeding approach described here may be useful in producing mice that model aspects of non-AD pathological aging, providing a useful comparison model to mice that exhibit pathological features more similar to AD and AD/LBD pathology. Ethics approval Human brain tissue was obtained from the Mayo Clinic Brain Bank and University of Florida Neuromedicine Human Brain Tissue Bank with informed consent following institutional regulations. All animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee. Consent for publication Not applicable. Availability of data and materials All data generated and analyzed during this study are included in this manuscript. Competing interests The authors declare that they have no competing interests. Funding This work was supported by funding from the National Institute of Health; grant numbers R21AG046125, R21AG055113, U54NS110435, P50AG047266, P30AG066506. Authors’ contributions Study concept and design: BDM, YL, JL, BIG, DRB. Pathological analysis: SP and DD. Acquisition of data: BDM, YL, GX, HH, MFA, CLC, HSF, CM, SF, SP, DD. Statistical analysis: BDM and DRB. Analysis and interpretation of the data: BDM, YL, GX, CJ, JL, BIG, TEG, DRB. Drafting of the manuscript: BDM and DRB. Acquisition of funding: JL, BIG, TEG, DRB. All authors read and approved the final manuscript. Acknowledgements The authors would like to thank the patients and their families that donated tissue. We thank Dr. Peter Davies (Feinstein Institute for Medical Research, Manhasset, NY, USA) posthumously for antibodies to tau. Conflict of interest TEG is a co-founder of Lacerta Therapeutics. References 1. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184-5. 2. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595-608. 3. Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487-91. 4. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409-21. 5. Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer's disease. Am J Pathol. 2008;173(3):762-72. 6. Guo Q, Li H, Cole AL, Hur JY, Li Y, Zheng H. Modeling Alzheimer's disease in mouse without mutant protein overexpression: cooperative and independent effects of Abeta and tau. PLoS One. 2013;8(11):e80706. 7. Angulo SL, Orman R, Neymotin SA, Liu L, Buitrago L, Cepeda-Prado E, et al. Tau and amyloid-related pathologies in the entorhinal cortex have divergent effects in the hippocampal circuit. Neurobiol Dis. 2017;108:261-76. 8. Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, et al. Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol. 2017;187(7):1601-12. 9. Rodriguez GA, Barrett GM, Duff KE, Hussaini SA. Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Abeta and tau pathology in the hippocampus. PLoS Biol. 2020;18(8):e3000851. 10. Saul A, Sprenger F, Bayer TA, Wirths O. Accelerated tau pathology with synaptic and neuronal loss in a novel triple transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34(11):2564-73. 11. Xu G, Ulm BS, Howard J, Fromholt SE, Lu Q, Lee BB, et al. TAPPing into the potential of inducible tau/APP transgenic mice. Neuropathol Appl Neurobiol. 2022;In Press. 12. Koller EJ, Ibanez KR, Vo Q, McFarland KN, Gonzalez De La Cruz E, Zobel L, et al. Combinatorial model of amyloid beta and tau reveals synergy between amyloid deposits and tangle formation. Neuropathol Appl Neurobiol. 2021;48(2):e12779. 13. Ulm BS, Borchelt DR, Moore BD. Remodeling Alzheimer-amyloidosis models by seeding. Mol Neurodegener. 2021;16(1):8. 14. Vergara C, Houben S, Suain V, Yilmaz Z, De Decker R, Vanden Dries V, et al. Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019;137(3):397-412. 15. Nies SH, Takahashi H, Herber CS, Huttner A, Chase A, Strittmatter SM. Spreading of Alzheimer tau seeds is enhanced by aging and template matching with limited impact of amyloid-beta. J Biol Chem. 2021;297(4):101159. 16. Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, et al. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40(1):1-8. 17. Perry RH, Irving D, Blessed G, Fairbairn A, Perry EK. Senile dementia of Lewy body type. A clinically and neuropathologically distinct form of Lewy body dementia in the elderly. J Neurol Sci. 1990;95(2):119-39. 18. Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281-9. 19. Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98(21):12245-50. 20. Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, et al. Amyloid-Beta (Abeta) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Abeta Pathology. Neuron. 2020;105(2):260-75 e6. 21. Lloyd GM, Dhillon JS, Gorion KM, Riffe C, Fromholt SE, Xia Y, et al. Collusion of alpha-Synuclein and Abeta aggravating co-morbidities in a novel prion-type mouse model. Mol Neurodegener. 2021;16(1):63. 22. Yamada M, Tsukagoshi H, Otomo E, Hayakawa M. Systemic amyloid deposition in old age and dementia of Alzheimer type: the relationship of brain amyloid to other amyloid. Acta Neuropathol. 1988;77(2):136-41. 23. Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer's disease: the need for a paradigm shift. Neuron. 2011;69(2):203-13. 24. Jack CR, Jr., Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132(Pt 5):1355-65. 25. Moore BD, Chakrabarty P, Levites Y, Kukar TL, Baine AM, Moroni T, et al. Overlapping profiles of Abeta peptides in the Alzheimer's disease and pathological aging brains. Alzheimers Res Ther. 2012;4(3):18. 26. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13(1):179-89. 27. Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16(3):285-98; discussion 98-304. 28. Golde TE, Dickson D, Hutton M. Filling the gaps in the abeta cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3(5):421-30. 29. Ayers JI, Fromholt S, Sinyavskaya O, Siemienski Z, Rosario AM, Li A, et al. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol Ther. 2015;23(1):53-62. 30. Kim JY, Grunke SD, Levites Y, Golde TE, Jankowsky JL. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J Vis Exp. 2014(91):51863. 31. Dhillon JS, Trejo-Lopez JA, Riffe C, Levites Y, Sacino AN, Borchelt DR, et al. Comparative analyses of the in vivo induction and transmission of alpha-synuclein pathology in transgenic mice by MSA brain lysate and recombinant alpha-synuclein fibrils. Acta Neuropathol Commun. 2019;7(1):80. 32. Saido TC, Kawashima S, Tani E, Yokota M. Up- and down-regulation of calpain inhibitor polypeptide, calpastatin, in postischemic hippocampus. Neurosci Lett. 1997;227(2):75-8. 33. Schmidt ML, DiDario AG, Otvos L, Jr., Hoshi N, Kant JA, Lee VM, et al. Plaque-associated neuronal proteins: a recurrent motif in neuritic amyloid deposits throughout diverse cortical areas of the Alzheimer's disease brain. Exp Neurol. 1994;130(2):311-22. 34. McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88-100. 35. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1-11. 36. Charidimou A, Martinez-Ramirez S, Shoamanesh A, Oliveira-Filho J, Frosch M, Vashkevich A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology. 2015;84(12):1206-12. 37. Xu G, Ran Y, Fromholt SE, Fu C, Yachnis AT, Golde TE, et al. Murine Abeta over-production produces diffuse and compact Alzheimer-type amyloid deposits. Acta Neuropathol Commun. 2015;3:72. 38. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476-81. 39. Strang KH, Goodwin MS, Riffe C, Moore BD, Chakrabarty P, Levites Y, et al. Generation and characterization of new monoclonal antibodies targeting the PHF1 and AT8 epitopes on human tau. Acta Neuropathol Commun. 2017;5(1):58. 40. Xia Y, Bell BM, Giasson BI. Tau K321/K353 pseudoacetylation within KXGS motifs regulates tau-microtubule interactions and inhibits aggregation. Sci Rep. 2021;11(1):17069. 41. Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34(4):521-33. 42. Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781-4. 43. Chakrabarty P, Rosario A, Cruz P, Siemienski Z, Ceballos-Diaz C, Crosby K, et al. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One. 2013;8(6):e67680. 44. Vallet PG, Guntern R, Hof PR, Golaz J, Delacourte A, Robakis NK, et al. A comparative study of histological and immunohistochemical methods for neurofibrillary tangles and senile plaques in Alzheimer's disease. Acta Neuropathol. 1992;83(2):170-8. 45. Hanna A, Iremonger K, Das P, Dickson D, Golde T, Janus C. Age-related increase in amyloid plaque burden is associated with impairment in conditioned fear memory in CRND8 mouse model of amyloidosis. Alzheimers Res Ther. 2012;4(3):21. 46. Chakrabarty P, Hudson VJ, III, Sacino AN, Brooks MM, D'Alton S, Lewis J, et al. Inefficient induction and spread of seeded tau pathology in P301L mouse model of tauopathy suggests inherent physiological barriers to transmission. Acta Neuropathol. 2015;130(2):303-5. 47. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25(4):402-5. 48. Xu G, Fromholt SE, Chakrabarty P, Zhu F, Liu X, Pace MC, et al. Diversity in Abeta deposit morphology and secondary proteome insolubility across models of Alzheimer-type amyloidosis. Acta Neuropathol Commun. 2020;8(1):43. 49. Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ. Exogenous induction of cerebral beta-amyloidosis in betaAPP-transgenic mice. Peptides. 2002;23(7):1241-7. 50. McKinley MP, DeArmond SJ, Torchia M, Mobley WC, Prusiner SB. Acceleration of scrapie in neonatal Syrian hamsters. Neurology. 1989;39(10):1319-24. 51. Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, et al. Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 2014;128(6):791-803. 52. Bidhendi EE, Bergh J, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T. Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J Clin Invest. 2016;126(6):2249-53. 53. Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, et al. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;106(31):12926-31. 54. Ziegler-Waldkirch S, d'Errico P, Sauer JF, Erny D, Savanthrapadian S, Loreth D, et al. Seed-induced Abeta deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer's disease. EMBO J. 2018;37(2):167-82. 55. Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci. 2011;31(41):14488-95. 56. Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83-95. 57. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909-13. 58. Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer's brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213(12):2635-54. 59. Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013;587(6):717-23. 60. Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129(2):221-37. 61. Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37(47):11406-23. 62. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110(23):9535-40. 63. Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136(Pt 4):1128-38. 64. Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75(3):351-62. 65. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, et al. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci. 2000;20(10):3606-11. 66. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330(6006):980-2. 67. Saha J, Dean DN, Dhakal S, Stockmal KA, Morgan SE, Dillon KD, et al. Biophysical characteristics of lipid-induced Abeta oligomers correlate to distinctive phenotypes in transgenic mice. FASEB J. 2021;35(2):e21318. 68. Preusser M, Strobel T, Gelpi E, Eiler M, Broessner G, Schmutzhard E, et al. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry. 2006;77(3):413-6. 69. Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247-50. 70. Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-beta deposition in vivo. Mol Psychiatry. 2012;17(12):1347-53. 71. Fritschi SK, Langer F, Kaeser SA, Maia LF, Portelius E, Pinotsi D, et al. Highly potent soluble amyloid-beta seeds in human Alzheimer brain but not cerebrospinal fluid. Brain. 2014;137(Pt 11):2909-15. 72. Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, et al. Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14(11):1017-22. 73. Eisele YS, Fritschi SK, Hamaguchi T, Obermuller U, Fuger P, Skodras A, et al. Multiple factors contribute to the peripheral induction of cerebral beta-amyloidosis. J Neurosci. 2014;34(31):10264-73. 74. Fritschi SK, Cintron A, Ye L, Mahler J, Buhler A, Baumann F, et al. Abeta seeds resist inactivation by formaldehyde. Acta Neuropathol. 2014;128(4):477-84. 75. Ye L, Rasmussen J, Kaeser SA, Marzesco AM, Obermuller U, Mahler J, et al. Abeta seeding potency peaks in the early stages of cerebral beta-amyloidosis. EMBO Rep. 2017;18(9):1536-44. 76. Kim J, Chakrabarty P, Hanna A, March A, Dickson DW, Borchelt DR, et al. Normal cognition in transgenic BRI2-Abeta mice. Mol Neurodegener. 2013;8:15.
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |