|
|
|
Free Neuropathology 2:34 (2021) |
|
Original Paper |
|
Friedreich cardiomyopathy is a desminopathy |
|
Arnulf H. Koeppen1,2, Rahman F. Rafique1, Joseph E. Mazurkiewicz3, Steven Pelech4,5, Catherine Sutter4, Qishan Lin6, Jiang Qian7 |
|
1 Research Service, Veterans Affairs Medical Center, Albany, NY, USA |
|
Corresponding author: |
|
Submitted: 11 November 2021 Accepted: 08 December 2021 Copyedited by: Vanessa S. Goodwill Published: 13 December 2021 |
|
Keywords: Friedreich ataxia, Cardiomyopathy, Proteomics, Desmin, αB-crystallin, Desminopathy |
|
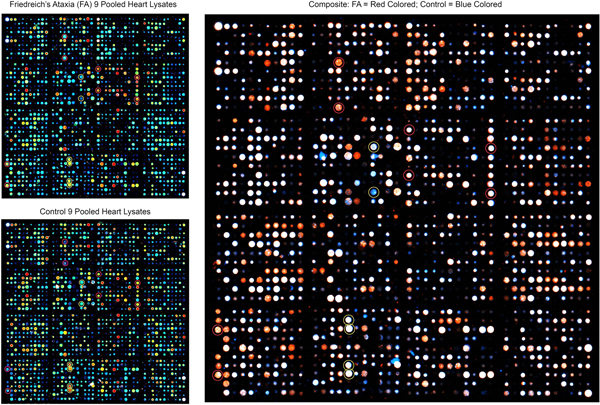
Abstract Heart disease is an integral part of Friedreich ataxia (FA) and the most common cause of death in this autosomal recessive disease. The result of the mutation is lack of frataxin, a small mitochondrial protein. The clinical and pathological phenotypes of FA are complex, involving brain, spinal cord, dorsal root ganglia, sensory nerves, heart, and endocrine pancreas. The hypothesis is that frataxin deficiency causes downstream changes in the proteome of the affected tissues, including the heart. A proteomic analysis of heart proteins in FA cardiomyopathy by antibody microarray, Western blots, immunohistochemistry, and double-label laser scanning confocal immunofluorescence microscopy revealed upregulation of desmin and its chaperone protein, αB-crystallin. In normal hearts, these two proteins are co-localized at intercalated discs and Z discs. In FA, desmin and αB-crystallin aggregate, causing chaotic modification of intercalated discs, clustering of mitochondria, and destruction of the contractile apparatus of cardiomyocytes. Western blots of tissue lysates in FA cardiomyopathy reveal a truncated desmin isoprotein that migrates at a lower molecular weight range than wild type desmin. While desmin and αB-crystallin are not mutated in FA, the accumulation of these proteins in FA hearts allows the conclusion that FA cardiomyopathy is a desminopathy akin to desmin myopathy of skeletal muscle. Introduction Heart disease in Friedreich ataxia (FA), hereafter called FA cardiomyopathy, is the most common cause of death in this otherwise rather neurological disease. FA cardiomyopathy was introduced as an integral part of FA by Russell (1946), and there is no doubt that the cardiac lesion is due to frataxin deficiency (Perdomini et al., 2014). Under the microscope, sections of FA heart show multiple abnormalities, including fiber hypertrophy (Koeppen et al., 2015), fiber necrosis, fibrosis, inflammatory infiltration, chaotic disorganization of intercalated discs, and accumulation of iron in a small percentage of cardiomyocytes without iron excess in the whole heart (Ramirez et al., 2012; Kruger et al., 2016). The reason for the highly variable pathological phenotype of FA as it affects heart, central and peripheral nervous systems, eyes, and endocrine pancreas has not been fully established. The current hypothesis is that structural and signaling proteins are affected downstream from cellular frataxin deficiency. On antibody microarrays and Western blots, desmin and its chaperone, αB-crystallin, are upregulated in FA cardiomyopathy. Desminopathy is well known from the study of desmin-related skeletal myopathies, in which cardiomyopathy is frequent (Dalakas et al., 2000; Goldfarb et al., 2004; Hnia et al., 2015). Desmin mutations cause aggregation in skeletal muscle, and some experiments have suggested that posttranslational modifications of desmin, such as phosphorylation and proteolytic fragmentation, make the protein amyloidogenic (Agnetti et al., 2014; Kedia et al., 2015). Based on the observations detailed in this report, the authors propose that FA cardiomyopathy is a protein aggregation myopathy involving desmin and αB-crystallin. In its BioGRID data base of protein, genetic, and chemical interactions (version 4.3.196), Uniprot (2021) lists 96 desmin-interacting proteins. Among them, αB-crystallin is prominently recognized as a desmin partner or “chaperone”. Indeed, αB-crystallin mutations cause “desmin” myopathy (Goldfarb and Dalakas, 2009). While desmin is an abundant cytoskeletal protein, it also has signaling properties, and the term “mechanochemical” may be applicable (Hnia et al., 2015). Material and methods Autopsy specimens: The Institutional Review Board of the Veterans Affairs Medical Center in Albany, NY, USA has approved this investigation. Heart autopsy specimens were obtained from 35 patients with FA through a national FA tissue repository in Albany, NY. Thirty-three patients had homozygous guanine-adenine-adenine (GAA) trinucleotide repeat expansions, and two were compound heterozygotes (Becker et al., 2017). Thirty-four patients had clinical heart disease or FA cardiomyopathy by histology. One FA patient had no heart disease during life or under the microscope. Normal hearts were made available by National Disease Research Interchange (Philadelphia, PA, USA). At the time of autopsy, a transverse slice weighing approximately 50 g, was made midway between the apex and atrioventricular groove, and immediately frozen at -20°C or, if available, at -80°C. The remainder of the heart was fixed in cold phosphate buffered formalin (pH 7.4). Samples of formalin-fixed left ventricular wall (LVW) were embedded in paraffin and sectioned at 6 µm for routine stains, immunohistochemistry, laser scanning confocal Congo Red fluorescence microscopy, and double-label immunofluorescence with antibodies to desmin and αB-crystallin, or desmin and ATP synthase F1 subunit β (ATP5B), a mitochondrial marker. Antibody microarray: The principles of this proteomic method, advantages, and pitfalls were described previously in detail (Yue and Pelech, 2018). Tissue samples were homogenized by sonication at ice temperatures in a lysis buffer containing (concentrations in parentheses) 3-morpholinopropane-1-sulfonic acid buffer pH 7.2 (20 mM), ethylene glycol-bis (β-aminoethyl ether), N,N,N’,N’-tetra acetic acid (2 mM), ethylenediaminetetraacetic acid (2 mM), sodium fluoride (50 mM), β-glycerophosphate (60 mM), sodium pyrophosphate (25 mM), sodium orthovanadate (5 mM), phenyl arsine phosphate (50 nM), Triton X-100 (1%), and sodium dodecylsulfate (0.05%). The lysis buffer also contained a protease inhibitor cocktail of aprotinin (0.5 µM), benzamidine (3 mM), 4(2-aminoethyl) benzene sulfonyl fluoride hydrochloric acid (1 mM), leupeptin (10 µM); and dithiothreitol (1 mM) to disrupt disulfide bonds. Protein concentrations were determined by Bradford’s method (1976). Proteins were cleaved at cysteine residues by the addition of tris (2-carboxyethyl) phosphine hydrochloride (10 mM) to the lysis buffer, followed by 2-nitro-5-thiocyanatobenzoic acid (100 mM) after sonication. Pooled lysates were biotinylated (Yue et al., 2017) and layered over 2 microarrays displaying, respectively, 900 and 1150 antibodies to a total of 2,050 antibodies (Kinexus Bioinformatics, Vancouver, BC, Canada). Proteins bound to the antibody spots were detected by incubation with a 1:1 mixture of Cy3- and Alexa 546-labeled anti-biotin. The fluorescent signals from the scanned microarrays were captured in a TIF file image. Signals were then quantified with ImaGene 9.0 microarray image analysis software (BioDiscovery, El Segundo, CA, USA) and normalized according to the sum of all the recorded signals per field. The values for duplicate measurements were averaged, and the mean value for the difference in duplicates from the average was typically ~10%. Processing lysates of FA and control hearts in triplicate allowed statistical determination of significant up- or down-regulation of proteins in FA by the Student t-test. Frataxin assay by enzyme-linked immunosorbent assay (ELISA): Details of frataxin ELISA were published before (Koeppen et al., 2015), but the amplifying steps with biotinylated anti-rabbit IgG and horse radish peroxidase-labeled streptavidin were omitted. The reason for the change was the presence of biotin in non-fat dry milk that was used as a suppressor protein. Briefly, heart lysates obtained by the method of Condò et al. (2006) were diluted 10-fold in phosphate-buffered saline (pH 7.2) (PBS) to reduce the concentration of nonionic detergents to less than 0.1%. The diluted lysates were centrifuged through membrane filter units with a molecular mass cutoff of 30 kDa (Millipore Sigma, Burlington, MA, USA). Wells of polystyrene ELISA plates were coated with a layer of monoclonal anti-frataxin (Abcam, Cambridge, MA, USA, cat. No. ab268063), followed by aliquots of filtered lysates (corresponding to 4 mg original tissue) or human recombinant frataxin (10 pg to 1 ng; Abcam, cat. No. ab95502). The sequence of subsequent additions (washing steps omitted) was polyclonal anti-frataxin (Abcam, cat. No. ab175402) and horseradish peroxidase-labeled goat anti-rabbit gamma globulin (Sigma, St. Louis, MO, USA, cat. No. A4914). The chromogenic solution contained ortho-phenylenediamine (2 mM) and 0.01% hydrogen peroxide in sodium phosphate-citric acid buffer (pH 5). Absorbances at 492 nm were determined in an ELISA plate reader manufactured by Molecular Devices (San José, CA, USA). Frataxin levels were expressed as ng/g wet weight. Coimmunoprecipitation: Pooled tissue lysate of 6 FA hearts, corresponding to 150 mg wet tissue in a volume of 150 µL, were mixed with 5 µg rabbit polyclonal anti-desmin and 50 µL of a slurry of protein A on Sepharose beads (Thermo Scientific, Rockford, IL, USA, cat. No. 29333). After overnight incubation at 4 ͦC, the Sepharose particles were collected by brief centrifugation and washed with phosphate-buffered saline (PBS). Bound proteins were released from protein A-Sepharose by Laemmli’s sample buffer and heating at 100 ͦC for 15 min. After cooling, the remaining Sepharose particles were precipitated by brief centrifugation, and 24 µL aliquots were electrophoresed on 10% polyacrylamide gels. At the end of the electrophoresis, proteins were transferred by electroblotting onto polyvinyledenefluoride (PVDF) membranes. The PVDF membranes were cut into longitudinal strips for the detection of established or putative desmin-associated proteins (frataxin, αB-crystallin; α-actinin, ankyrin 1, α-spectrin, plectin, and ZASP [cypher]). Sources of antibodies were as follows: anti-desmin (polyclonal, Invitrogen, Rockford, IL, USA. cat No. PA5-16705); anti-αB-crystallin (monoclonal, Santa Cruz Biotechnology, Dallas, TX, USA. cat. No. sc-137129); anti-α-actinin (monoclonal, Abcam, Waltham, MA, USA, cat. No. AB62298); anti-ankyrin 1 (monoclonal, Santa Cruz Biotechnology, cat. No. sc-12719); anti-α-spectrin (monoclonal, Santa Cruz, cat No. sc-48382); anti-plectin (monoclonal, Santa Cruz Biotechnology, cat. No. 37649); anti-ZASP (cypher) (monoclonal, Santa Cruz Biotechnology, cat. No. sc-374359); and anti-frataxin (monoclonal, Abcam, Branford, CT, USA; cat. No. ab268063). Mass spectrometry: Protein bands of interest on Coomassie Blue-stained SDS-PAGE gels were manually excised and minced. The pieces were dehydrated with acetonitrile for 10 min, vacuum dried, rehydrated with 5 mM triphosphine hydrochloride in 25 mM ammonium bicarbonate (pH 8.5) at 37 °C for 1 h and then alkylated with 100 mM iodoacetamide in 25 mM ammonium bicarbonate (pH 8.5) at room temperature for 1 h. The pieces were washed twice with 50% acetonitrile in 25 mM ammonium bicarbonate (pH 8.5) for 15 min, dehydrated with acetonitrile for 10 min, dried, and digested in sequencing grade modified trypsin (25 ng/µL; Sigma, St. Louis, MO, USA, cat. No. T8658) in 25 mM ammonium bicarbonate (pH 8.5) at 37 °C overnight. Following digestion, tryptic peptides were extracted three times with 50% acetonitrile containing 5% formic acid for 15 min each time while being vortexed. The extracted solutions were pooled and evaporated to dryness under vacuum. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed on an integrated micro-LC-Orbitrap Velos system (Thermo), comprising a Waters CapLC microscale chromatography system (Waters Corp., Milford, MA) with an auto-sampler, a stream-select module configured for pre-column plus analytical capillary column, and an Orbitrap Velos mass spectrometer fitted with an H-electrospray ionizer probe operated under Xcalibur 2.2 control. Injected samples (30 µL) were first trapped and desalted isocratically on a home-packed C18 pre-column cartridge (Everest C18, 5 µm particle diameter; column dimensions: internal diameter, 500 µm; length, 15 mm [Grace, Deerfield, IL]) for 6 min with 0.1% formic acid delivered by an auxiliary pump at 40 μL/min. The peptides were then eluted from the pre-column and separated on an ACE C18 capillary column (15 cm x 500 μm internal diameter) packed with ACE C18-300 particles (5 µm resin, Advanced Chromatography Technologies, Aberdeen, Scotland). The C18 column was connected in-line with the mass spectrometer and eluted at a flow rate of 20 µL/min with a 40-min gradient of 5-to-80% acetonitrile in 0.1% formic acid. Tandem spectrum data was processed using Mascot 2.7 (Matrix Science, Boston, MA). The lists of peaks were used to query a human protein database downloaded from National Center of Biotechnology Information by setting the following parameters: peptide mass tolerance, 10 ppm; MS/MS ion mass tolerance, 0.1 Da; allowing up to one missed cleavage; considering variable modifications such as methionine oxidation, cysteine carboxyamidomethylation, and deamidation. Only significant hits as defined by Mascot probability analysis was considered for a positive protein identification. Immunohistochemistry: To visualize desmin, its chaperone protein αB-crystallin, and known desmin-linked intermediate filament proteins (α-spectrin; ankyrin-1; plectin; ZASP [cypher]; and α-actinin), 6-µm thick paraffin sections were rehydrated by sequential incubation in a series of xylene, absolute ethanol, 95% ethanol, 80% ethanol, and deionized water. Endogenous peroxidase activity was suppressed by incubation in methanol containing 3% aqueous hydrogen peroxide. Antigen retrieval, background suppression, incubation with polyclonal or monoclonal antibodies, secondary biotinylated anti-rabbit IgG (for rabbit polyclonal primary antibodies) or anti-mouse IgG (for primary mouse monoclonal antibodies), and chromogenic solutions of diaminobenzidine and hydrogen peroxide were as described (Koeppen et al., 2020). Double-label fluorescence and immunofluorescence: To examine possible amyloidogenicity of desmin in FA cardiomyopathy, immunofluorescence of desmin and fluorescence of Congo Red were combined. Slide processing for immunofluorescence was as described before (Koeppen et al., 2020), and the fluorophore for desmin was Alexa Fluor 488-labeled goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA, cat. No. 115-545-003). After washing, the sections were incubated in a solution of high pH Congo Red, as described by the supplier (IHC World, Woodstock, MD, USA), washed in water, covered by PBS containing 50% glycerol (by vol), and cover-slipped. Excitation by laser light at 488 nm was suitable for the fluorescence of both Alexa Fluor 488 (desmin) and Congo Red. Images were collected in a Zeiss LSM 880 laser scanning confocal microscope (Oberkochen, Germany) set to a band pass of 507-544 nm for Alexa Fluor 488 and 560-600 nm for Congo Red. Double-label immunofluorescence of the pairs desmin/αB crystallin and desmin/mitochondrial ATP5B followed a published protocol (Koeppen et al., 2020). ATP5B, the β subunit of ATP synthase F1, was detected by a polyclonal antibody (Santa Cruz Biotechnology, Dallas, TX, USA, cat. No. sc 33618) and Cy3-labeled secondary goat anti-rabbit IgG (Jackson ImmunoResearch, cat. No. 111-165-003). Results Antibody microarray of heart lysates: Each dot in the panels of figure 1 represents a unique validated antibody, and the legend describes how fluorescent signals of FA and controls were superimposed and analyzed. Thirteen proteins were upregulated to variable degrees on the 900-antibody microarray. When expressed as percent change from control (%CFC), the following proteins stood out: adducin, %CFC 210; αB-crystallin, %CFC 194; insulin receptor substrate, %CFC 324; serine/threonine kinase type 4, %CFC 103; ribosomal protein S kinase alpha 3, %CFC 149. The 1150-antibody microarray of Kinexus (Yue, et al, 2018) yielded modest upregulation of 23 other proteins that were not further analyzed. The up-regulation of αB-crystallin prompted a more detailed study of this protein and its partner, desmin, by SDS-PAGE, Western blots, mass spectrometry, and double-label immunofluorescence (figs. 2-8).
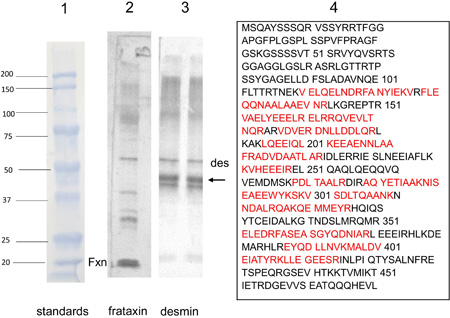
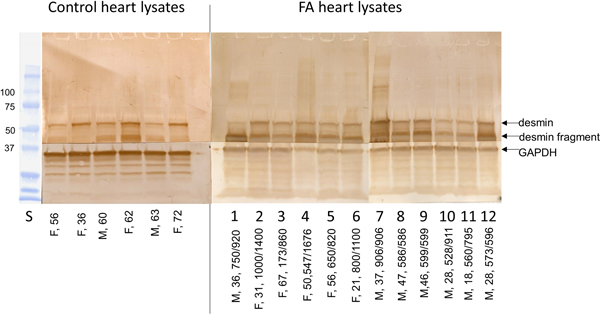
Figure 1. Antibody microarray of pooled heart lysates of 9 FA patients (left upper panel) and 9 normal controls (left lower panel). In the smaller panels, pseudocolors represent high fluorescence (red) and lower fluorescence (blue). Intermediate fluorescence intensity is represented by green and yellow, in that order. The larger image was created by overlaying the FA and control images (FA on top). All spots of the control image were made blue, and all FA spots were made red. Using Adobe Photoshop technology, the FA images on top were rendered 50% transparent so that the bottom spots are white. If FA intensity was higher than that of the matching control, the spot appears as red. If the composite signal is blue, the signal in FA was lower than that of the control. Pooling of lysates was intended to control for between-samples variability, and performance in triplicate allowed statistical analysis by t-testing. The next step in the experiment was SDS-PAGE and Western blotting of proteins in pooled and individual lysates. Frataxin levels: The frataxin level in 12 FA heart lysates (mean ± standard deviation) was 23.3±3.5 ng/g wet weight. In 4 control heart lysates the level was 522.5 ± 35 ng/g wet weight. The difference was significant at p=0.022. Western blots: Figure 2 shows a Western blot of desmin and frataxin in pooled FA heart lysates. The pooled lysates represent equal additions of the first 6 FA cases shown in figure 3. The polyclonal antibody to desmin revealed a low intensity band at 53 kDa which represents the canonical protein. The strongest desmin signal occurred from a fragment of approximately 45 kDa. Mass spectrometric analysis of this fragment (fig. 2) confirmed its derivation from desmin. While pooling of 12 heart lysates was useful in the control of inter-sample variability in FA, the Western blot in figure 3 shows that the desmin fragment is shared by all FA samples. The qualitative intensity of the desmin band did not correlate with the length of the GAA trinucleotide repeat expansions in the shorter allele of the frataxin gene (fig. 3). A comparable desmin isoprotein is also present in one normal control (fig. 2, left panel, subject F,62).
Figure 2. Western blot of desmin and frataxin in 6 pooled FA heart lysates and mass spectrometry of a proteolytic desmin fragment. Lane 1: Prestained proteins of known mass. Lane 2: Western blot of frataxin (Fxn). Pooling of heart lysates allows the detection of residual frataxin in FA. Lane 3: Western blot of desmin. The canonical desmin migrates at 53 kDa (des), but a second, strongly reactive desmin band (arrow) is located at approximately 45 kDa. Lane 4: Mass spectrometry of the truncated desmin fragment in lane 3 (arrow). The amino acids in red were identified by liquid chromatography-tandem mass spectrometry. The sequences represent signature peptides of desmin.
Figure 3. Western blots of desmin in 6 normal control and 12 FA heart lysates. All wells received a total protein load of 50 µg. The arrows show the location of canonical desmin at 53 kDa, a desmin fragment, and GAPDH (as a loading control). The PVDF membranes were cut horizontally to generate two segments of the blot and allow visualization of GAPDH. Sex and ages are shown for the lanes of control lysates (left); in the right panel, of FA lysates, the entries include sex, age, and GAA trinucleotide repeats. In 8 of 12 FA lysates, the blots show prominent expression of a proteolytic desmin fragments at approximately 45 kDa.
Figure 4. Desmin co-immunoprecipitation. Desmin in pooled lysates of 6 FA hearts was precipitated by anti-desmin, and the protein-antibody complex was recovered on protein A Sepharose. Desmin and other proteins were released from protein A by heating in Laemmli’s sample buffer containing β-mercaptoethanol. Aliquots were electrophoresed by SDS-PAGE, electroblotted onto PVDF membranes, and visualized by immunochemistry with antibodies to the stated proteins. The Western blot shows desmin bands at approximately 53 kDa and between 40 and 45 kDa, respectively. A distinct band of αB-crystallin is also present. Other lighter bands at 45 kDa, 25 kDa and 15 kDa are considered preparative artifacts. Immunohistochemistry: In normal hearts, antibodies to desmin and αB-crystallin revealed reaction product in intercalated discs and Z-discs. Anti-αB-crystallin also showed M bands on longitudinal sections of heart fibers that were desmin negative (fig. 5f). In FA, desmin and αB-crystallin reaction product showed abnormally wide, thickened, and hyperconvoluted intercalated discs and aggregates in the sarcoplasm (fig. 5). Desmin and αB-crystallin reaction product in the two compound heterozygous FA cases showed a similar degree of protein aggregate myopathy. Reaction product for β-spectrin, ankyrin, plectin, and ZASP (cypher) was less informative, though α-spectrin immunohistochemistry displayed the chaotic disorganization of intercalated discs. α-Actinin reaction product showed the striation of cardiomyocytes by reacting with Z-discs but was not useful to assess the state of intercalated discs.
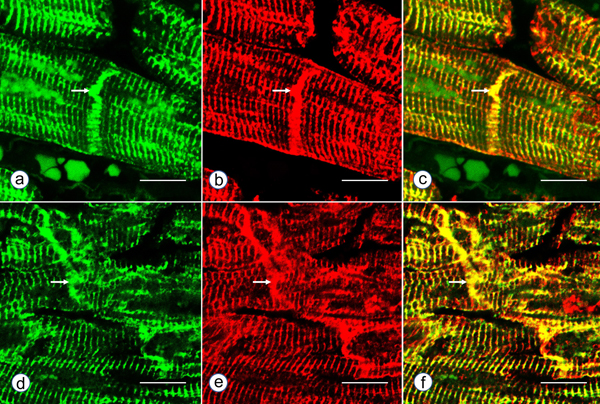
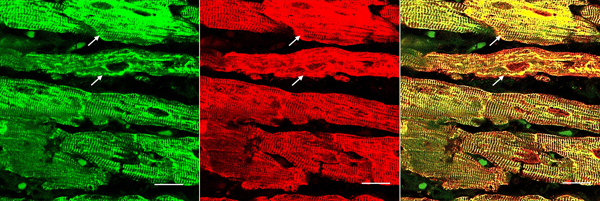
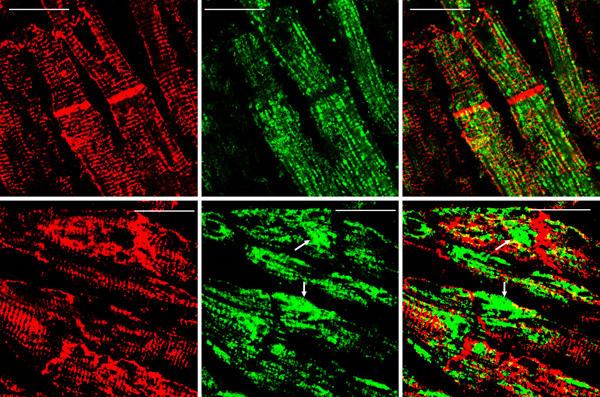
Figure 5. Immunohistochemistry of desmin and αB-crystallin in left ventricular wall. a-b, desmin; c-f, αB-crystallin. a and c, normal left ventricular wall; b, d-f, FA. In normal myocardium, desmin (a) and αB-crystallin (c) are localized in intercalated and Z discs. In FA, desmin reaction product reveals accumulation in or near disorganized intercalated discs (b, arrow). αB-crystallin immunohistochemistry shows wide and fragmented intercalated discs (d and e). Enlargement of the square in e displays αB-crystallin reaction product in M bands (f, arrow, M). M bands are desmin-negative. Bars; a-e, 10 µm; f, 3 µm. FA patients: b, F, 67 GAA 621/766; d-f, F, 69, GAA 560/560. Double-label laser scanning confocal immunofluorescence: Figure 6 shows the colocalization of desmin and αB-crystallin in Z-discs and intercalated discs. In FA, intercalated discs are wider at approximately 50 µm than normal (approximately 23 µm) and have undergone chaotic disorganization (fig. 6d). A more advanced fiber destruction is illustrated in figure 7. Two adjacent fibers display accumulation of desmin and αB-crystallin, obliterating the contractile apparatus of the cardiomyocytes. A search for amyloid by polarizing light for apple-green birefringence and Congo Red fluorescence did not reveal amyloid deposits. Figure 8 shows double-label immunofluorescence of desmin and ATP5B. All fibers in FA contain clusters of mitochondria, visualized by green fluorescence of ATP5B, and one cluster is surrounded by a rim of desmin. In normal cardiomyocytes, mitochondria occur in rows of delicate granules between heart fibrils (fig. 8, top row).
Figure 6. Double-label laser scanning confocal immunofluorescence microscopy of desmin and αB-crystallin. Desmin immunofluorescence (a, d) is Alexa Fluor 488 green, αB-crystallin (b,e) Cy3 red. Merged images appear in c and f. The top panel (a-c) represents a normal control, the bottom panel (d-f) FA (man, 24, GAA 700/1050). Desmin and αB-crystallin fluorescence colocalize at intercalated discs (arrows) and Z discs. In FA, an intercalated disc has undergone chaotic modification. Bars, 10 µm.
Figure 7. Desmin and αB-crystallin overload in FA cardiomyopathy (same patient as in fig. 6). Desmin is shown as Alexa Fluor 488 green, αB-crystallin as Cy3 red, The merged image on the right shows extensive colocalization of these protein and destructive aggregation in two fibers (arrows). Bars, 10 μm.
Figure 8. Double-label laser scanning confocal immunofluorescence microscopy of desmin and ATP5B. Top row, normal control; bottom row, FA (same patient as in fig. 5). Desmin is shown as Cy3 red, ATP5B as Alexa Fluor 488 green. In the normal control, mitochondria appear as rows of punctate green organelles aligned along fibrils. In FA, green mitochondria appear in clusters (arrows). The upper cluster is surrounded by a desmin deposit. Bars, 20 µm. Discussion Frataxin levels in FA hearts: The detection limit of the described ELISA is 10 pg. We followed biochemical tradition by expressing frataxin levels as mass per gram wet weight, which is at variance with the current practice of FA investigators to express frataxin as ng/total protein. The data must be interpreted with some caution because we assumed that the wet weight of normal and FA heart tissues was close to its water content. Tissue lysates contained inhibitory substances that interfered with ELISA. Filtration through membranes with a molecular mass cutoff of 30 kDa removed the interference but may have introduced an additional error in the calculation of corresponding wet weight. Desmin in FA cardiomyopathy: Uniprot (2021) identifies 36 natural variants of human desmin, of which some are pathogenic mutations. None are listed that could account for the deletion of an estimated 80 amino acids that would explain the high signal desmin isoprotein of 45 kDa in FA hearts (figs. 2 and 3). Clemen et al. (2008) attributed pathogenicity in patients with skeletal muscle desminopathy to the presence of a low-abundance truncated desmin. It is unknown whether this pathological mechanism is analogous to the prominent desmin band on our Western blots of FA patients (fig.3). Desmin is not mutated in FA, and FA patients have no skeletal myopathy. Arbustini et al. (1998) presented Western blots of heart and muscle lysates from patients with desmin-related cardiomyopathy that showed two isoforms of desmin of about the same intensity at masses of 55 and 53 kDa, respectively. A truncated muscle desmin of the cases of skeletal myopathy reported by Clemen et al. (2008) migrated at 54 kDa, rather than the 45 kDa in the lysates of FA hearts reported here (figs. 2 and 3). We assume that the truncated desmin in FA cardiomyopathy derives from normal desmin by proteolysis (fig. 2). The functional role of desmin-containing intermediate filaments in skeletal and cardiac muscle is complex. Authors who published three-dimensional renditions of desmin in skeletal muscle (Goldfarb and Dalakas, 2009; Clemen et al., 2013) and heart muscle (Capetanaki et al., 2015) emphasized the localization of the protein at Z discs and intercalated discs (heart only), but also near the nuclear and sarcoplasmic membranes, and surrounding mitochondria. The role of αB-crystallin in possible proteolysis and aggregation of desmin in FA cardiomyopathy is uncertain, but αB-crystallin is viewed as essential for the proper assembly of mature desmin filaments (Ghosh et al., 2007; Sharma et al., 2017). Peculiarly, mutations of αB-crystallin cause myopathy with a phenotype that is very similar to desmin myopathy, including desmin accumulation in muscle tissue (Goldfarb et al., 2004). Autosomal dominant mutations in the desmin gene cause desmin myopathy that may be followed by cardiomyopathy later in the course of the disease. In contrast, the desmin cardiomyopathy reported here is not followed by skeletal myopathy. Desmin and mitochondria in FA cardiomyopathy: Frataxin is a mitochondrial protein, and FA is a mitochondrial disease. Figure 8 illustrates the clustering of mitochondria near desmin aggregates, but it is unknown whether this seeming entrapment impairs mitochondrial function in FA. References Agnetti G, Halperin VL, Kirek JA, Chakire K, Guo YJ, Lund L, Nicolini F, Gherli T, Guarnieri C, Caldarera CM, Tomaselli GF, Kass DA, Van Eyk JE. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc Res 2014; 102:24-34. https://doi.org/10.1093/cvr/cvu003 Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, Bellini O, Dal Bello B, Campana C, Piccolo G, Febo O, Opasich C, Gabazzi A, Ferrans VJ. Restrictive cardiomyopathy, atrioventricular block and mild subclinical myopathy in patients with desmin-immunoractive material deposits. J Am Coll Cardiol 1998; 3:645-53. https://doi.org/10.1016/s0735-1097(98)00026-6 Becker AB, Qian J, Gelman BB, Yang M, Bauer P, Koeppen AH. Heart and nervous system pathology in compound heterozygous Friedreich ataxia. J Neuropathol Exp Neurol 2017; 76: 665-75. https://doi.org/10.1093/jnen/nlx047 Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72:348-54. https://doi.org/10.1006/abio.1976.9999 Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatswellas G, Tsikitis M. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol 2015; 32:113-20. https://doi.org/10.1016/j.ceb.2015.01.004 Clemen CS, Fischer D, Heimann J, Eichinger L, Müller CR, Müller HD, Goebel HH, Schröder R. How much mutant protein is needed to cause a protein aggregate myopathy in vivo? Lessons from an exceptional desminopathy. Hum Mutat 2008; 30:E490-E499. https://doi.org/10.1002/humu.20941 Clemen CS, Herrmann H, Strelkov SV, Schröder R. Desminopathies: pathology and mechanisms. Acta Neuropathol 2013; 125:47-75. https://doi.org/10.1007/s00401-012-1057-6 Condò I, Ventura N, Malisan F, Tomassini B, Testi RE. A pool of extramitochondrial frataxin that promotes cell survival. J Biol Chem 2006; 281:16750-6. https://doi.org/10.1074/jbc.M511960200 Dalakas, MC, Park K-Y, Semino-Mora C, Lee, HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. New Engl J Med 2000; 342:770-80. https://doi.org/10.1056/NEJM200003163421104 Ghosh JG, Houck SA, Clark JI. Interactive sequences in the stress protein and molecular chaperone human αB crystallin recognize and modulate the assembly of filaments. Int J Biochem Cell Biol 2007; 39:1804-15. https://doi.org/10.1016/j.biocel.2007.04.027 Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain 2004; 127(Pt 4):723-34. https://doi.org/10.1093/brain/awh033 Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009; 119:1806-13. https://doi.org/10.1172/JCI38027 Hnia K, Rumspacher C, Vermot J, Laporte J. Desmin in muscle and associated disease: beyond the structural function. Cell Tiss Res 2015; 360:591-608. https://doi.org/10.1007/s00441-014-2016-4 Kedia N, Arhzaouy K, Pittman SK, Sun Y, Batchelor M, Weihl CC, Bieschke J. Desmin forms toxic, seeding-competent amyloid aggregates that persist in muscle fibers. Proc Natl Acad Sci USA 2019; 116(34):16835-4. https://doi.org/10.1073/pnas.1908263116 Koeppen AH, Ramirez RL, Becker AB, Bjork ST, Levi S, Santambrogio P, Parsons PJ, Kruger PC, Yang KX, Feustel PJ, Mazurkiewicz JE. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One 2015; 10:e116396. https://doi.org/10.1371/journal.pone.0116396 Koeppen AH, Qian J, Travis AM, Sossei AB, Feustel PJ, Mazurkiewicz JE. Microvascular pathology in Friedreich cardiomyopathy. Histol Histopathol 2020; 35:39-46. https://doi.org/10.14670/HH-18-132 Kruger PC, Yang KX, Parsons PJ, Becker AB, Feustel PJ, Koeppen AH. Abundance and significance of iron, zinc, copper, and calcium in the hearts of patients with Friedreich ataxia. Am J Cardiol 2016; 18:127-31. https://doi.org/10.1016/j.amjcard.2016.04.024 Perdomini M, Belbellaa B, Monassier L, Reutenauer L, Messaddeq N, Cartier N, Crystal REG, Aubourg P, Puccio H. Prevention, and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat Med 2014; 20:542-7. https://doi.org/10.1038/nm.3510 Ramirez RL, Qian J, Santambrogio P, Levi S, Koeppen AH. Relation of cytosolic iron excess to the cardiomyopathy of Friedreich's ataxia. Am J Cardiol 2012; 110:1820-7. https://doi.org/10.1016/j.amjcard.2012.08.018 Russell DS. Myocarditis in Friedreich’s ataxia. J Path Bact 1946; 63:739-48. https://doi.org/10.1002/path.1700580414 Sharma S, Conover GM, Elliott JL, Der Perng M, Herrmann H, Quinlan RA. αB-crystallin is a sensor for assembly intermediates and for the subunit topology of desmin intermediate filaments. Cell Stress Chaperones 2017; 22:613-26. https://doi.org/10.1007/s12192-017-0788-7 Uniprot: the universal protein knowledge base. Nucleic Acids Res 2021; 49(D1):D480-D489. https://doi.org/10.1093/nar/gkaa1100 Uniprot. BioGRID Biological General Repository for Interaction Datasets 2021. https://doi.org/10.25504/FAIRsharing.9d5f5r Yue L, Sam C, Arora N, Winkler DFH, Pelech S. Antibody microarray and immunoblotting analyses of the EGF signaling phosphorylation network in human A431 epidermoid carcinoma cells. Clin Proteom Bioinformat 2017; 2:1-10. https://doi.org/10.15761/CPB.1000119 Yue L, Pelech S. Application of high content antibody microarrays for biomarker discovery and tracking cellular signaling. Adv Proteom Bioinformat 2018:1-24. https://doi.org/10.29011/APBI-107.100007
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |