|
|
|
Free Neuropathology 2:15 (2021) |
|
Opinion Piece |
|
Viral infection and dementia: A brief synthesis |
|
Clayton A. Wiley |
|
Division of Neuropathology, Department of Pathology, University of Pittsburgh, Pittsburgh, PA, USA |
|
Corresponding author: |
|
Submitted: 17 May 2021 Accepted: 4 June 2021 Copyedited by: Shino Magaki Published: 8 June 2021 |
|
Keywords: Dementia, Emergent infections, Immunosenescence, Neurodegeneration, Syphilis, Viral encephalitis |
|
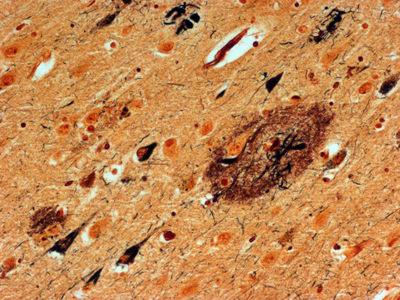
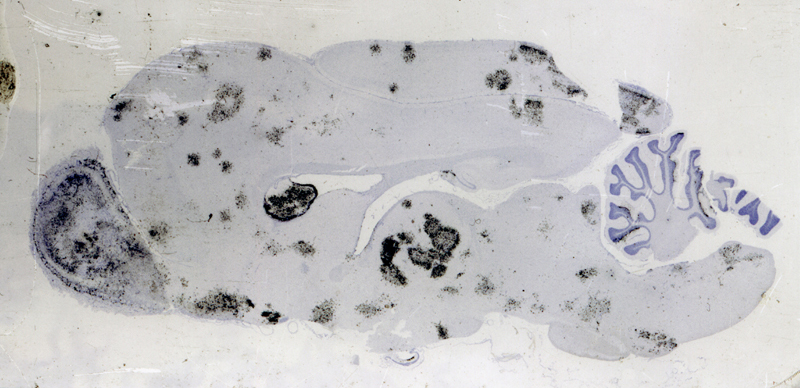
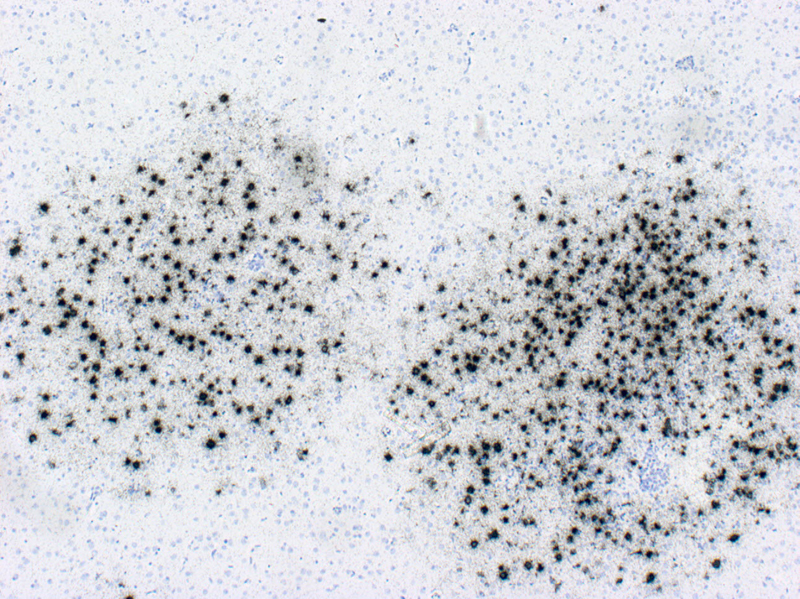
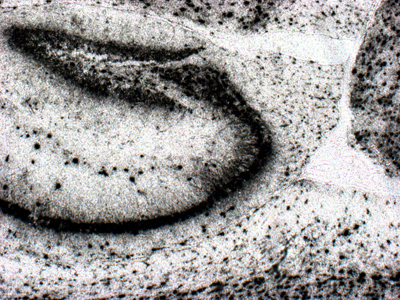
Abstract For the past 400 years, the most common cause of dementia was tertiary syphilis [1]. Its prevalence declined dramatically with the advent of potent antibiotics in the 20th century, but these same antibiotics also helped increase our average lifespan, leading to dramatic increases in the prevalence of age-related dementias. Abundant progress has been made connecting early onset dementias with mutations in neural genes. Late onset dementias have been linked to a more enigmatic set of genes, some of which have been connected to neuroinflammation, begging the question: Are age-related dementias linked to infection? Numerous studies have reported an association between dementia and infections in general and viral infections in particular. While these associations have been subject to extensive reviews, the purpose of this synthesis is to examine the hypothesized link of viral infections and dementia from the opposite perspective: What do we know about acute and chronic encephalitides that could forge a link with dementias? There appears to be little support for the concept that viral infections are a major contributor to today’s common dementias. However, the emergence of new central nervous system (CNS) viral infections, coupled with senescent immune and nervous systems in our aged population, create new opportunities for infections to contribute to dementia. Introduction There may be as many clinical definitions for dementia as there are pathological definitions for neurodegeneration. A simple clinical definition of dementia might be “a chronic irreversible encephalopathy affecting multiple cognitive domains” (memory being the most prominently involved domain in the most common form of dementia, Alzheimer’s disease [AD]). Clinical dementias can be measured with a variety of clinical scales but are most commonly appreciated from the perspective of impaired capacity to perform activities of daily living. A simple pathological definition of neurodegeneration could encompass loss of neurons, their axonal connections or synapses, but given difficulties in quantifying these degenerative changes, current pathological diagnoses rely on distinctive histopathology (Figure 1) [2, 3]. With recent refinements in our understanding of dementia and neurodegeneration, is there support for an infectious etiology in age-related dementias?
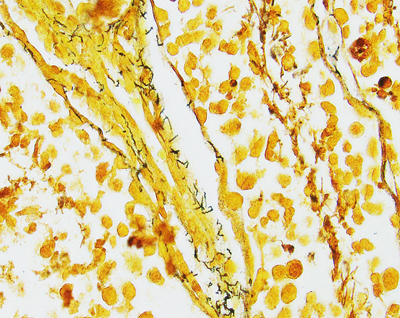
Figure 1. Bielschowsky-stained section of neocortex from a patient with Alzheimer’s disease. Individual pyramidal neurons show intense staining of neurofibrillary tangles in cell bodies and proximal processes. Neuropil plaques show staining of dystrophic neuritic processes. There are no infiltrating adaptive immune cells. Syphilis: The most common form of dementia It has been hypothesized that syphilis was brought to Europe with the return of various European conquistadors [1]. In the old-world, syphilis spread mercilessly infecting up to 20% of the population, 5% of whom eventually developed dementia [4]. So prevalent was tertiary syphilis that Sir William Osler held it as the paradigmatic neurologic disease saying, “Know paretic neurosyphilis in all aspects and you know all of psychiatry.” [5] By the beginning of the 20th century tertiary syphilis was the most common form of dementia [4]. Fortunately, the advent of antibiotics in the 20th century coupled with the exquisite sensitivity of Treponema pallidum, led to rapid decline in prevalence and incidence of infection and commensurate decrease in syphilitic dementia. Which is not to say syphilis is gone. Today it infects 0.5% of the world’s population with 6 million new infections per year (~100,000 in North America). The agent Treponema pallidum (Figure 2) causes a protean complex of diseases of which its tertiary form, mediates dementia. Without antibiotic therapy, syphilis can establish a lifelong infection that evades and exhausts the immune system. Tertiary syphilis is the result of years of organism replication to the point that unfettered growth throughout the brain along with macrophage activation causes a global chronic encephalitis with neurodegeneration and dementia [5]. It is an unusual form of encephalitis, distinct from the acute viral infections described below, with little evidence of cell mediated immunity and instead a remarkable abundance of activated microglia in gray matter regions of abundant T. pallidum. As with most infections, perivascular infiltrates are prominent and with the spirochetal tropism for vascular walls can progress to an end arteritis infarctive syndrome. Histopathologic changes are widespread with minimal evidence of a system specific distribution except for a frontal/temporal cortical predominance. There is extensive neuronal loss and reactive astrocytosis but no intraneuronal protein aggregates.
Figure 2A. Warthin Starry stain of rabbit testes infected with Treponema pallidum. Short thin corkscrew like organisms (black) are seen adjacent to a small blood vessel.
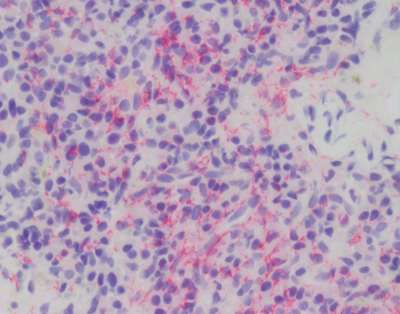
Figure 2B. Immunostain (red) for T. pallidum shows a dense collection of microorganisms. Age-related dementia Perversely, the cure of the most common cause of infectious dementia was followed years later by an increasing prevalence of age-related dementias that plague us today. The same antibiotic development that fueled elimination of syphilis also protected people from common pathogens and thus (along with improvements in sanitation) indirectly increased our average life span. Prior to the introduction of agriculture, the average life span of Homo sapiens has been estimated to be less than 35 years [6]. With agriculture and the industrial revolution, by 1900 the average lifespan increased in the US to 47 years. Add in modern sanitation and medicine and by 1950 the average lifespan was 66 for males and 71 for females. A mere 70 years later the average bumped another decade to 76 and 81 for males and females, respectively. Unfortunately, one thing we know about dementia today with absolute certainty is, it is age dependent. The Alzheimer’s Association estimates that in the US, one in 10 people over the age of 65 has AD (one of the most common forms of dementia) (https://www.alz.org/alzheimers-dementia/facts-figures, accessed 12.15.20). Despite many of those dying 3-5 years after diagnosis, by 85 years of age 30% of the population is demented. Said another way, syphilitic dementia disappeared and was more than replaced by age-related dementia. Is there evidence that this age-related dementia is related to an infectious agent? Infection-related dementia There have been scattered but intense investigations to discover an infectious etiology for today’s common dementias. These studies have spanned simple attempts to morphologically identify an infectious agent within the brains of affected individuals, to sophisticated and sensitive metagenomics sequencing studies. Numerous reports, some of which were replicated, have identified a gamut of bacterial or viral agents in the brains of deceased previously demented individuals [7]. However, given the prevalence of dementia and the abundance of opportunities to find an associated infectious agent, it is difficult to imagine that a prevalent direct etiologic agent has been missed. That conclusion should be stated with some diffidence given the famous example of Helicobacter pylori linked to gastric and duodenal ulcers [8], but it is a fair summary to say, no infectious agent has been consistently associated with common forms of age-related dementia known today. But is presence of the infectious agent within the brain tissue the sine qua non linking an agent to dementia? It is theoretically possible that an infectious agent could initiate the neurodegenerative process or mediate it remotely without directly invading the brain. For example, direct infection of neurons perturbs cellular metabolism that could potentially disrupt normal protein synthesis or proteasome/autophagy degradation leading to accumulation of aggregated proteins and neuronal dysfunction. Indirect effects of systemic infection on neurophysiology could include decreased perfusion secondary to general cytokine induced blood coagulation or altered neuronal gene expression induced by specific cytokines like interferons. This brief review will be limited to assessing potential direct viral etiologies of human dementias from the perspective of what we know about acute and chronic viral encephalitis that could link them to dementias. The reader is referred to a selection of reviews assessing potential of bacterial etiologies [7]. Prions (proteinaceous infectious particles) which are infectious agents known to cause dementia will also not be discussed because they are exquisitely rare, part of the host genome rather than a viral agent and have been the subject of many comprehensive reviews [9]. Why do we care about viruses? Our biosphere has evolved from competition amongst replicating agents. Uni- and multicellular hosts have evolved numerous defenses to ward off invasion by infectious agents. This dynamic state persists with continuous evolution in host defense and agent offense. Viruses are one class of infectious agents. Perhaps most succinctly defined by Peter Medawar as a piece of bad news wrapped in a protein [10], viruses have evolved to co-opt the host cell energy and machinery to replicate the viral genome. The strategies for accomplishing this are legion and in rare cases involve infecting the host brain. How do viruses infect the brain? Viral infection can occur through a variety of pathways [11]. To move from infected to naïve host, viruses must traverse the environment and access the surface of living cells which, in the case of droplet and aerosol transmission, implies contact with a mucous membrane, but in the case of arthropod born viruses, is the result of direct subcutaneous injection. At the cell surface, specific host membrane ligands bind viral proteins vastly facilitating viral entry. Intracellular replication increases the number of infectious particles (virion) that can disseminate to sites for a secondary amplification (frequently in lymphoid structures) before greater dissemination throughout the host (e.g., the brain). Viral entry into a host cell immediately triggers the host cell’s innate immunity, launching a race between the virus and host immune system that ends with either cessation of viral replication and clearance of the virus, or a variety of debilitating outcomes for the host. Upon cell entry cytoplasmic and membrane bound host proteins of the innate immune system immediately detect viral proteins and nucleic acids initiating a cascade of antiviral machinery (e.g., interferon) to block viral replication in the infected and adjacent cells. Unfortunately, evolution has selected for an array of viral mechanisms to evade the host innate immune response. The partial thwarting of explosive viral replication gives the host’s second line of defense, adaptive immunity, a chance to mount a complex humoral and cellular response. Adaptive immunity takes precious days to develop, so if the virus gains entry to the CNS during that window, it may gain a temporal advantage over the growing adaptive immune response resulting in a costly destructive battle inside the CNS. The CNS is protected from viral invasion by an elaborate system of physical, chemical and physiological barriers [12]. Encased within the cranial vault, direct introduction of a viral agent is essentially impossible, so viruses take two other routes to enter the brain: hematogenous or transaxonal. Both of these other routes require initially establishing an infection in the periphery. Hematogenous dissemination requires that the virus reach a concentration (titer) in the blood that can broach the blood brain barrier (BBB) by passage through, or infection of, endothelial cells. An intriguing variation on this route is viral entry into white blood cells and transit through the BBB hidden within a monocyte Trojan Horse. A second route, transaxonal, has been perfected by many viruses and requires infection of peripheral nerves that naturally extend cellular processes into the brain, such that the virus can utilize intracellular transport mechanisms to rapidly enter the CNS sanctuary. Viral encephalitis Viruses that successfully circumvent peripheral innate immunity and access the CNS through pathways described above, have the opportunity to encounter CNS cells [11]. If viral surface proteins bind to host neuroglial membrane receptors, the virus is taken into the cell where it can be detected by innate immune machinery similar to that in the periphery. Neuroglial elements may not be able to unleash the same potent anti-viral innate immunity as peripheral cells. Within a closed chamber the brain can tolerate only limited cytotoxic edema. Additionally, given exquisite balance of RNA processing within the CNS, it may not tolerate potent interferon related arrest of RNA metabolism. Like other organs, the brain has histiocytes (microglia) that are specialized in detecting pathogens and attracting peripheral immune surveillance [13]. As adaptive immunity is brought to bear on infected CNS cells, cellular destruction leads to neurological dysfunction which is accentuated by cytotoxic edema. Simple brain edema associated with immune attack is enough to cause neurological dysfunction and through swelling, brain herniation and death. Add to that the viral and cytotoxic effector arms of the immune system and it is not surprising that encephalopathy is the clinical outcome. Viral-host duals display a spectrum of histopathologies. Rapidly growing viruses with the capacity to lyse cells combined with robust immune responses can lead to frank necrosis, while less dramatic infectious battles result in non-necrotic inflammatory reactions [14]. Most viral encephalitides are acute infections ending with either host death or viral clearance achieved in a matter of days. Chronic viral encephalitis is a more nuanced outcome, characterized by persistent adaptive immune response in the context of continued viral replication. Do acute viral encephalitides lead to dementia? Viruses mediating acute encephalitis cover a wide range of families but are mostly RNA viruses. Because of their temporal time course and systemic symptoms, acute viral encephalitides are not likely to be confused clinically with a progressive dementia. As the host immune response brings viral replication under control, cerebral edema can mediate global neural dysfunction, clinically manifest as delirium. With clearance of virus and abatement of the immune response, a brain lesion resulting from the inflammatory process can manifest as a static neurologic deficit, but the clinical picture is not one of a progressive neurodegenerative disease. Firstly, the time course of acute encephalitis is in days not months to years. Secondly, distribution of infection within the CNS, with rare exception, is global rather than system specific as seen with most dementias. Thirdly, with the exception of immune compromised hosts, acute encephalitis pathology demonstrates a severe inflammatory response rather than subtle neurodegenerative changes with pathognomonic aggregated proteins (Figure 3).
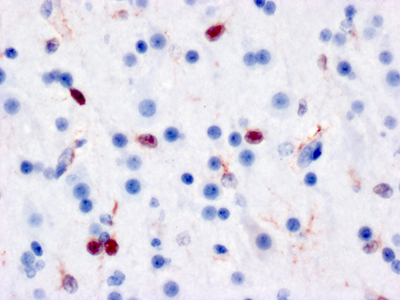
Figure 3A. Immunostain for CD3 of neocortical tissue from the brain of a child who died of Coxsackie viral encephalomyocarditis shows dense infiltration of lymphocytes (red).
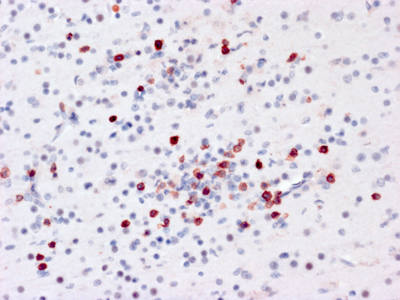
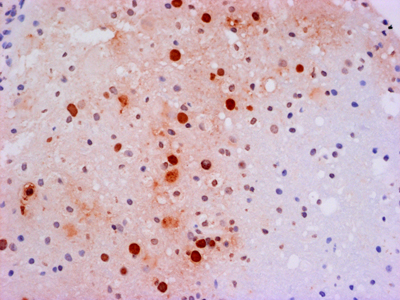
Figure 3B. Immunostain for Coxsackie viral antigens shows staining of multiple neuroglial cells in both the nucleus and cytoplasmic processes. Do chronic viral encephalitides lead to dementia? The spectrum of viruses mediating chronic encephalitis is more limited and has proportionally more representation from DNA viruses. Some chronic encephalitides (e.g., herpes simplex virus [HSV]) are the end product of millennia of co-evolution between host and virus. While HSV can cause an acute encephalitis in the immunologically compromised, in the immune intact host HSV has achieved a unique latent infection in the peripheral nervous system (PNS) where viral synthesis is shut down and re-awakened in times of stress permitting active disease and transmission. Regardless of organ, chronic infection results from a dynamic balance where the virus does not kill the host and the host does not develop an adequate immune response that can eradicate infection. In addition to outflanking the innate immune response, many viruses have developed the means to mediate some level of general or specific immunosuppression. If this down regulation occurs prior to viral eradication, it could result in viral persistence with or without some low level of immune reaction. Alternatively, rather than causing immunosuppression themselves, viruses can take advantage of genetic or iatrogenic compromises of the host immune system. Finally like all other organs (including the brain), the immune system undergoes a natural senescence [15]. With age, stem cell elements dissipate and immunological memory fades. Loss of immunological surveillance can result in selective holes in adaptive immunity permitting chronic viral infections (e.g., progressive multifocal leukoencephalopathy, Figure 4).
Figure 4. In situ hybridization (red) for JC viral nucleic acids in the brain of a patient succumbing to progressive multifocal leukoencephalopathy. Individual oligodendroglial nuclei are filled with viral nucleic acid while surrounding parenchyma shows no T-cell infiltration. Potential chronic viral infections associated with dementia An example of a rare chronic viral encephalitis seen in children and young adults can be caused by two different viruses: rubeola and rubella. Childhood infection with these agents is usually followed by a self-limiting systemic disease marked by a distinctive skin rash. In rare children, for unknown reasons, but hypothesized to be related to an aberrant immune response, chronic viral infection persists in the CNS [16]. Unlike acute viral encephalitis, the immune system only dampens exponential viral growth, without destroying infected host cells to eradicate the virus. For years viral replication and a low-grade immune response compromise neuronal function and lead to atrophy and a global encephalopathy. Pathologically these diseases show muted inflammatory changes. HSV: Of perhaps all chronic viral infections potentially associated with neurodegeneration, herpesviruses may be the most studied. Herpesviridae are ancient viruses that have co-evolved with many mammalian species. Of the 8 known human herpesviruses, HSV is the most notorious for mediating neurological disease. In the fetus, newborn and immunocompromised, HSV mediates an acute and frequently necrotic pan-encephalitis. For unknown reasons, rare apparently immunologically intact individuals also develop an acute necrotizing encephalitis predominantly focused in the frontal and temporal lobes. While readily treatable, there are case reports where this acute infection transitions into a smoldering chronic infection, raising the prospects that it could be associated with a chronic neurodegenerative disease. The literature on this topic is immense and controversial with proponents on both sides of the debate for and against HSV involvement in neurodegeneration (reviews [17, 18]). While there is no consensus, the cumulative evidence would suggest that while low levels of HSV are detectable in CNS and PNS (latent infection), there is minimal to no evidence that active HSV replication is temporally or spatially associated with common neurodegenerative diseases. Whether HSV infection could initiate a cascade of inflammatory events that subsequently drives a degenerative process awaits construction of a pathogenically logical and testable hypothesis. SARS-CoV-2: In 2021 it is almost impossible to write anything without mentioning COVID-19. Much heat and little light has been published on the effect of SARS-CoV-2 infection on the nervous system. As with many severe infections, a variety of neurological signs and symptoms have been observed in COVID patients [19-21]. Observations of olfactory neuroepithelial infection [22] have led to the conjecture that SARS-CoV-2 enters the CNS by ascending axonal connections to the olfactory bulb. Such a dissemination has been demonstrated in transgenic murine models expressing human angiotensin converting enzyme 2 (ACE2; the human viral receptor) under a keratin promoter [23]. But this convenient animal system does not seem to actually model the human disease [24]. Intensive attempts have been made to identify SARS-CoV-2 infection in human autopsy tissue to little avail. The preponderance of evidence would suggest a non-direct infection effect on the nervous system such as overwhelming systemic immune activation (cytokine storm) leading to dissolution of the BBB and compromised CNS perfusion. In addition to these effects of acute infection, more chronic CNS symptoms have been documented in a subset of patients recovering from COVID (long-haulers). While early in our studies of COVID pathology, clearly this disease is open to the same theories that have developed with HSV associated neurodegeneration. The challenge will be to convert these theories into testable hypotheses. Is HIV the infectious dementia of our generation? Before AIDS was even proven to be caused by a viral infection, it was clearly associated with severe neurologic disease. Beyond the devastating opportunistic infections related to severe immunosuppression, late-stage AIDS patients also exhibited a unique dementia. In retrospect, the histopathology of AIDS dementia was exactly what should have been expected for a macrophage tropic virus in a patient with severe immunosuppression, minimal to no lymphocyte infiltration in the context of abundant virus [25] (Figure 5). Indeed, the concentration of virus in the CNS exceeded that in lymphoid organs [26]. The big mystery that has yet to be solved is, How did infection of microglia in the absence of neuroglial infection lead to a clinical dementia [27]? It has been hypothesized that infected microglia either produced a neurotoxin (e.g., quinolinic acid [28]) or were unable to carry out critical physiological functions that non-infected microglia normally perform (e.g., synaptic stripping). Hypotheses range from remote or local responses to immunologic stimulation to hypothesized abortive infection of astrocytes. Before intensive investigation could elucidate the pathogenesis of AIDS dementia, highly effective anti-retroviral therapy eradicated HIV encephalitis. While HIV infected individuals still experience neurologic symptoms of unknown etiology, AIDS dementia, like syphilis dementia, disappeared.
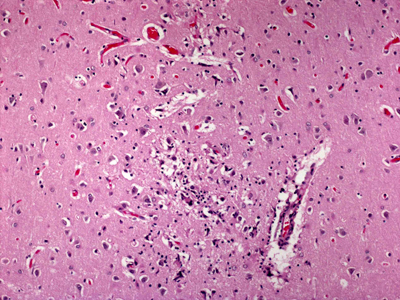
Figure 5A. Low power H&E-stained section of cortical tissue from a patient with HIV encephalitis.
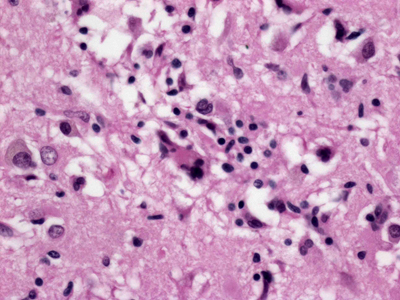
Figure 5B. Higher power H&E-stained section showing a multinucleated giant cell. What would make a dementia virus? With what we have learned from infections of the nervous system, what would be the characteristics of a host-viral interaction that would result in a neurodegenerative disorder of dementia? The abundance of RNA viruses that infect the brain would suggest that something about their biology makes them particularly effective at replication in the CNS. To effectively infect CNS cells, the hypothetical dementia virus would need a viral coat protein capable of binding host CNS cell membranes. Through its intricate specialization and regionalization there are an abundance of potential CNS region and system specific target proteins (e.g., neurotransmitter receptors). Infection of CNS cells would not necessarily lead to lysis of neuroglial elements but would incapacitate normal cell physiology leading to dysfunction or degeneration. Competition between normal host CNS RNA metabolism, viral RNA metabolism and innate immune responses could mimic many of the neurodegenerative mechanisms being investigated in neurodegenerative disorders like amyotrophic lateral sclerosis [29]. Alternatively, as seen with syphilis and HIV, infection of microglia might indirectly mediate neuronal dysfunction [27, 30]. How would the hypothetical virus reach the CNS and propagate between hosts? While many pathways could be used, aerosol transmission is the most effective means of spreading between high density host populations (Figure 6). Additionally, for entirely mysterious reasons, aerosol dissemination is highly effective at transferring virus to the brain. Viruses that normally are limited to peripheral infections, when aerosolized, lead to severe pan-CNS infection (e.g., Rift Valley fever virus, Figure 7).
Figure 6A. In situ hybridization autoradiography (black grains) for Influenza A in the brain of a ferret 8 days after aerosol exposure to H5N1. Numerous foci throughout the brain demonstrate abundant flu infected cells.
Figure 6B. Higher power image showing regions of intense neuroglial infection. But the most important feature of the hypothetical dementia virus infection is that it would occur in the context of immunosuppression. Many viruses elicit general or specific immunosuppression, with HIV notable for a particularly severe CD4 T-cell suppression. However, it is also possible that rather than causing immunosuppression on its own, the hypothetical virus could take advantage of senescence of the human immune system accompanying the aging of our population. With greater than 20% of our population over the age of 65 by 2050, there is no shortage of potential hosts. The COVID-19 pandemic has taught us the cost of not being prepared to combat emergent infections. The highly effective mRNA vaccines are the result of decades of intensive investigation. We need to broaden and deepen our understanding of how vaccination can be employed to confer protection of the brain particularly from aerosol infection in the immunosenescent host.
Figure 7. When Rift Valley fever virus (RVFV) is transmitted by mosquito bite, infection does not cause an encephalitis. However, when delivered as an aerosol, RVFV causes a global encephalitis. In situ hybridization autoradiography (black grains) for RVFV in the brain of a mouse 7 days after aerosol exposure to RVFV. Essentially all of the neurons are infected. Conclusion Historically infectious agents have been the primary cause of dementia. Introduction of antibiotics eradicated syphilitic dementia while at the same time helping humans live longer allowing them to develop even more prevalent age-related dementias. The recent emergence of HIV has once again proved the potential for an infectious agent to mediate a dementing illness. Fortunately, we discovered pharmaceuticals to arrest HIV infection and block development of immunosuppression and AIDS dementia. In the context of an increasingly aged population, living with senescent immune and nervous systems offers up new targets for emergent viral infections to wreak havoc on our cognitive capacities. However, for the most common age-related neurodegenerative diseases (e.g., AD, Lewy body disease, frontotemporal dementia) there is little in the histopathology defining these disorders to connect them to a viral etiology. References [1] Tampa M, Sarbu I, Matei C, Benea V, Georgescu SR. Brief history of syphilis. J Med Life. 2014;7:4-10. [2] Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1-11. [3] Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1-13. [4] Merritt HH, Adams RD, Solomon HC. Neurosyphilis. New York: Oxford University Press; 1946. [5] Ghanem KG. Evaluation and management of syphilis in the HIV-infected patient. Curr Infect Dis Rep. 2010;12:140-6. [6] Pahl KP. Life expectancy in ancient and modern man. Acta Anthropogenet. 1981;5:119-28. [7] Honjo K, van Reekum R, Verhoeff NP. Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement. 2009;5:348-60. [8] de Brito BB, da Silva FAF, Soares AS, Pereira VA, Santos MLC, Sampaio MM, et al. Pathogenesis and clinical management of Helicobacter pylori gastric infection. World J Gastroenterol. 2019;25:5578-89. [9] Walker LC. Prion-like mechanisms in Alzheimer disease. Handb Clin Neurol. 2018;153:303-19. [10] Medawar PB, Medawar JS. Aristotle to zoos: a philosophical dictionary of biology. Cambridge: Harvard University Press; 1985. [11] Johnson RT. Viral infections of the nervous system. 2nd ed. Philadelphia: Lippincott-Raven; 1998. [12] Johnson RT. Neurovirology: evolution of a new discipline. J Neurovirol. 1995;1:2-4. [13] Kofler J, Wiley CA. Microglia: key innate immune cells of the brain. Toxicol Pathol. 2011;39:103-14. [14] Love S, Budka H, Ironside JW, Perry A eds. Greenfield's neuropathology. 9th ed. Boca Raton: CRC Press; 2015. [15] Fukushima Y, Minato N, Hattori M. The impact of senescence-associated T cells on immunosenescence and age-related disorders. Inflamm Regen. 2018;38:24. [16] Townsend JJ, Baringer JR, Wolinsky JS, Malamud N, Mednick JP, Panitch HS, et al. Progressive rubella panencephalitis. Late onset after congenital rubella. N Engl J Med. 1975;292:990-3. [17] Itzhaki RF. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer's disease. Front Aging Neurosci. 2018;10:324. [18] Allnutt MA, Johnson K, Bennett DA, Connor SM, Troncoso JC, Pletnikova O, et al. Human herpesvirus 6 detection in Alzheimer's disease cases and controls across multiple cohorts. Neuron. 2020;105:1027-35 e2. [19] Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020;143:3104-20. [20] Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a Review. JAMA Neurol. 2020;77:1018-27. [21] Berlit P, Bosel J, Gahn G, Isenmann S, Meuth SG, Nolte CH, et al. "Neurological manifestations of COVID-19" - guideline of the German society of neurology. Neurol Res Pract. 2020;2:51. [22] Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2021;24:168-75. [23] Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. 2021;218:e20202135. [24] Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 2020;19:919-29. [25] Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci U S A. 1986;83:7089-93. [26] Pang S, Koyanagi Y, Miles S, Wiley C, Vinters HV, Chen IS. High levels of unintegrated HIV-1 DNA in brain tissue of AIDS dementia patients. Nature. 1990;343:85-9. [27] Wang T, Rumbaugh JA, Nath A. Viruses and the brain: from inflammation to dementia. Clin Sci (Lond). 2006;110:393-407. [28] Valle M, Price RW, Nilsson A, Heyes M, Verotta D. CSF quinolinic acid levels are determined by local HIV infection: cross-sectional analysis and modelling of dynamics following antiretroviral therapy. Brain. 2004;127:1047-60. [29] Butti Z, Patten SA. RNA dysregulation in amyotrophic lateral sclerosis. Front Genet. 2018;9:712. [30] McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9:14.
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |