|
|
|
Free Neuropathology 2:14 (2021) |
|
Case Report |
|
Desmoplastic myxoid tumor of pineal region, SMARCB1-mutant, in young adult |
|
Branavan Manoranjan1, Yves P. Starreveld1, Robert A. Nordal2, Christopher Dunham3, Susanne Bens4, Christian Thomas5, Martin Hasselblatt5, Jeffrey T. Joseph6 |
|
1 Department of Clinical Neurosciences, Division of Neurosurgery, University of Calgary, Canada |
|
Corresponding author: |
|
Submitted: 21 April 2021 Accepted: 23 May 2021 Copyedited by: Bert M. Verheijen Published: 01 June 2021 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Desmoplastic myxoid tumor, SMARCB1-mutant, Atypical teratoid/rhabdoid tumor, Pineal, CSF dissemination |
|
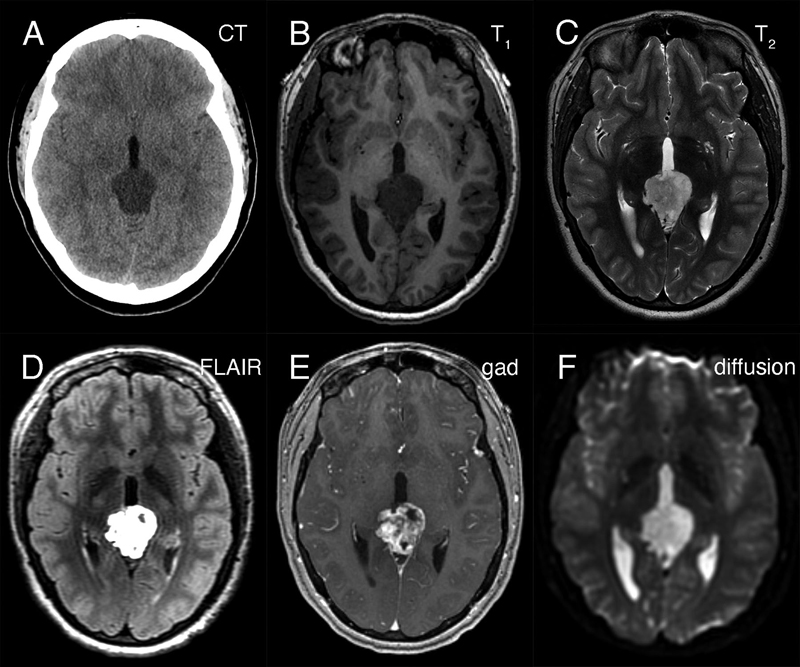
Abstract We present a young adult woman who developed a myxoid tumor of the pineal region having a SMARCB1 mutation, which was phenotypically similar to the recently described desmoplastic myxoid, SMARCB1-mutant tumor of the pineal region (DMT-SMARCB1). The 24-year-old woman presented with headaches, nausea, and emesis. Neuroimaging identified a hypodense lesion in CT scans that was T1-hypointense, hyperintense in both T2-weighted and FLAIR MRI scans, and displayed gadolinium enhancement. The resected tumor had an abundant, Alcian-blue positive myxoid matrix with interspersed, non-neoplastic neuropil-glial-vascular elements. It immunoreacted with CD34 and individual cells for EMA. Immunohistochemistry revealed loss of nuclear INI1 expression by the myxoid component but its retention in the vascular elements. Molecular analyses identified a SMARCB1 deletion and DNA methylation studies showed that this tumor grouped together with the recently described DMT-SMARCB1. A cerebrospinal fluid cytologic preparation had several cells morphologically similar to those in routine and electron microscopy. We briefly discuss the correlation of the pathology with the radiology and how this tumor compares with other SMARCB1-mutant tumors of the nervous system. Case Presentation The patient is a 24-year-old woman who presented following 2-weeks of progressive headache, nausea, emesis, and diplopia. Her medical history was significant for right sided hearing loss that began with tinnitus in 2014. Neurological examination revealed bilateral grade I papilledema but no other focal motor or sensory deficits. Preoperative non-contrast head computed tomography (CT) revealed a hypodense pineal region lesion extending into the posterior third ventricle and causing obstructive hydrocephalus (figure 1A). Magnetic resonance imaging (MRI) demonstrated a 3.0 x 3.4 x 2.6 cm mass in the pineal region (see below). Beta-human chorionic gonadotropin (beta-hCG) and alpha-fetoprotein (AFP) from serum and cerebrospinal fluid (CSF) were within normal limits. A spinal MRI did not identify visible leptomeningeal disease; however, CSF cytology had atypical cells (see below).
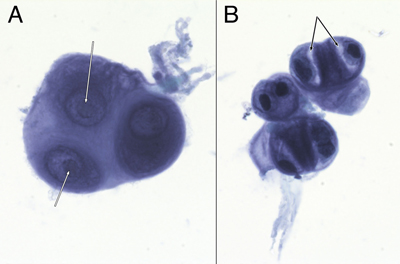
Figure 1: Preoperative neuroimaging. The tumor in the pineal region was hypodense on non-contrast CT head (A), hypointense in T1-weighted (B) and hyperintense in T2-weighted (C) MRI sequences. It was strongly hyperintense in FLAIR sequences (D), showed heterogeneous gadolinium enhancement (E), and did not demonstrate restricted diffusion (F). She underwent a successful endoscopic third ventriculostomy (ETV) and biopsy of the pineal lesion on post-admit day 3. The initial biopsy did not identify a germ cell tumor or a lymphoma, so the patient underwent tumor resection via a suboccipital craniotomy. Grossly, the tumor was opaque, grey-white in color, firm, and moderately vascular. The tumor was debulked, however it could not be completely resected where its superior edge was adherent to the vein of Galen and the basal vein of Rosenthal, and where it was adherent to the pulvinar. Post-operative MRI showed nodular enhancement along the right lateral margin of the surgical cavity. She was discharged home on postoperative day 8 but subsequently developed a pseudomeningocele that was treated with a programmable ventriculoperitoneal shunt. She remains well on follow-up at 5 months post-shunt insertion. Because of the CSF cytologic results (see below), she received craniospinal irradiation. Curative intent craniospinal irradiation with a radiation boost to the primary tumor region was prescribed. 36 Gy was delivered to the neuraxis in 1.8 Gy fractions, and an additional 19.8 Gy was given to the primary tumor volume. At last follow-up, five months after surgery, the patient remained well. Neuroradiology Preoperative non-contrast computed tomography (CT) revealed a hypodense pineal region tumor extending into the posterior third ventricle and causing obstructive hydrocephalus (figure 1A). Magnetic resonance imaging (MRI) showed that this mass was hypointense in T1-weighted images (figure 1B), hyperintense in T2-weighted (figure 1C) and FLAIR images (figure 1D). The mass heterogeneously enhanced with gadolinium (figure 1E) and was in close proximity to the deep cerebral veins, including the vein of Galen. It did not show diffusion restriction (figure 1F; ADC map not shown). No leptomeningeal disease or drop metastases were demonstrable on spinal MRI (data not shown). These indicated that the tumor was likely sparsely cellular, its matrix contained abundant but not free water, and it had vessels that lacked a blood-brain barrier. CSF Cytology The patient's initial CSF cytology (figure 2), which was obtained from a lumbar puncture before surgical intervention, included several small clusters of atypical cells that had a high nuclear-to-cytoplasmic ratio, prominent nucleoli (white arrows in 2A), and variable amounts of cytoplasm. Some cells had small zones of clear cytoplasm (black arrows in 2B) that were interpreted as vacuoles, while others lacked this feature (cell cluster in 2A). These cells were morphologically similar to those in the routine sections (figure 3F) and in the electron microscopy from the later tumor resection (see figure 6). Although these cells had some features of chondrocytes (see Arya, 2019; Chen, 1990; Takeda, 1981; Bigner, 1981; and the chapter on CSF cytology by Cibas, 2014), they showed increased pleomorphism and displayed considerably less cytoplasm than previously illustrated chondrocytes in CSF and lacked other debris that was illustrated in Arya, 2019.
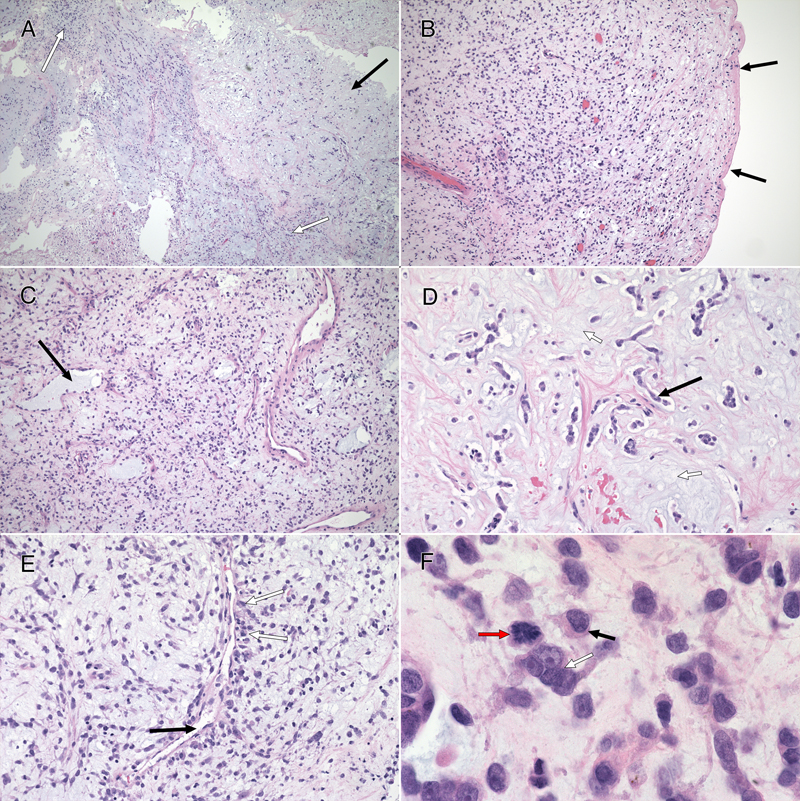
Figure 2: CSF Cytology. These cells from the preoperative CSF, imaged under oil at 100x magnification, were stained with toluidine blue. They had large nuclei with prominent nucleoli (A, white arrows). Some appeared multinucleated (A and B). Their cytoplasm showed moderate staining cytoplasm (A), although several cells had partial cytoplasmic clearing or vacuoles (B, black arrows). Tumor Histology The tumor had two distinct features: a major myxoid component (figure 3) and a minor, intermixed, vascular component that was associated with neuropil and astrocytes (figure 5, see below). The former had sparsely (figure 3, black arrow in A and D) to moderately cellular regions (white arrow in 3A) containing occasional microcysts (3C, black arrow) that were delineated by a single layer of cells. Although fragmented, the tumor had regions displaying a clear collagenous capsule (3B, black arrows). Because this capsule was not adherent to other tissues, it likely indicated that the tumour grew into a fluid-filled space, either within the third ventricle or into a meningeal cistern. In more myxoid, less cellular areas, the tumor cells formed small clusters or chains of cells (3D, black arrow) that were embedded in the loose, myxoid stroma (3D, white arrow). Fine eosinophilic strands meandered through the background stroma (3, D and E). Although these regions were histologically reminiscent of chordoma, the tumor did not express brachyury (data not shown). Tumor cells displayed a minor predilection for the perivascular region (3E, black arrow), although they did not form ependymal pseudorosettes or pilomyxoid rosettes. They often grew along barely visible eosinophilic strands (swirling pattern in 3E, chains of nuclei in 3D). At high magnification, the cells had small amounts of often eccentric, eosinophilic cytoplasm (3F, black arrow) and large, hyperchromatic nuclei. Many cells appeared binucleated or multinucleated (3F, white arrow) These were morphologically similar to the few available cells in the CSF cytology (compare with cells in figure 2). Mitoses were present (3F, red arrow) but infrequent (less than one per ten high-power fields) and the Ki67 proliferation index was 9% (see here).
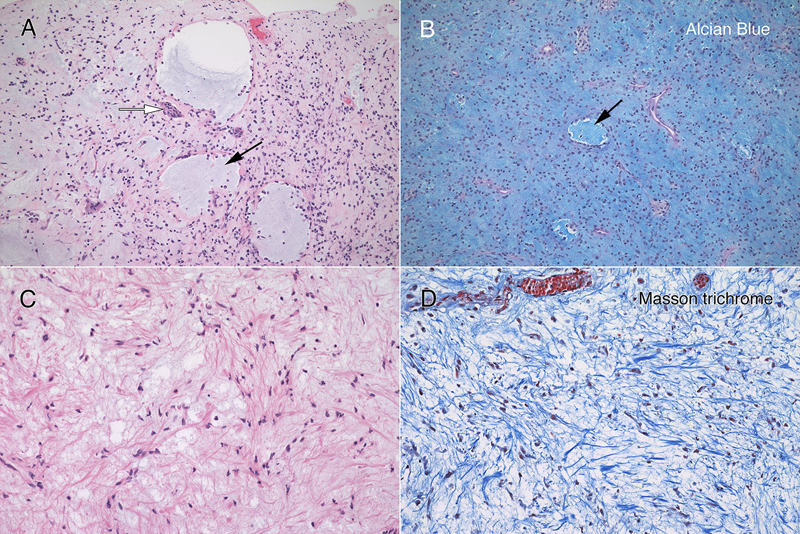
Figure 3: Routine Histology. All panels stained with hematoxylin-eosin. The resection (A; 4x) had hypocellular (black arrow) and moderately cellular (white arrows) areas. In some fragments (B; 10x), the tumor was delimited by an outer collagenous layer (black arrows). More densely cellular regions (C; 10x) had microcysts (black arrow) lined by single cells. A loose, hypocellular area (D; 20x) displayed chain-like growth (black arrow), with cells set in a myxoid matrix having fine, eosinophilic strands. Tumor cells showed a weak predilection for the perivascular region (E, white arrows; 20x) around thin-walled vessels (black arrow). At high magnification in F (oil immersion, 100x), the tumor cells had scant amount of often eccentric, eosinophilic cytoplasm (black arrow) and occurred in scattered small groups (white arrow). The red arrow highlights a mitotic figure. Clicking the picture will lead you to the full virtual slide (H&E). The tumor's myxoid matrix and its scattered microcysts (figure 4A) diffusely and strongly stained with Alcian blue (figure 4B), which indicates that the tumor elaborated a ground substance containing carbohydrate moieties. This myxoid matrix was pervasive throughout the tumor and was only absent in non-tumor vascular elements (see pink-staining vessels and islands in 4B). Unlike similar tumors, this neoplasm did not display an overt desmoplasia in the surrounding parenchyma (see figure 3). However, the tumor did produce an extensive network of fine, eosinophilic strands (see figure 3D and 4C), which stained strongly blue in Masson trichrome stains (figure 4D). This finding indicates that the tumor created an intrinsic desmoplastic collagenous stroma and likely explains why this myxoid tumor felt firm during surgery.
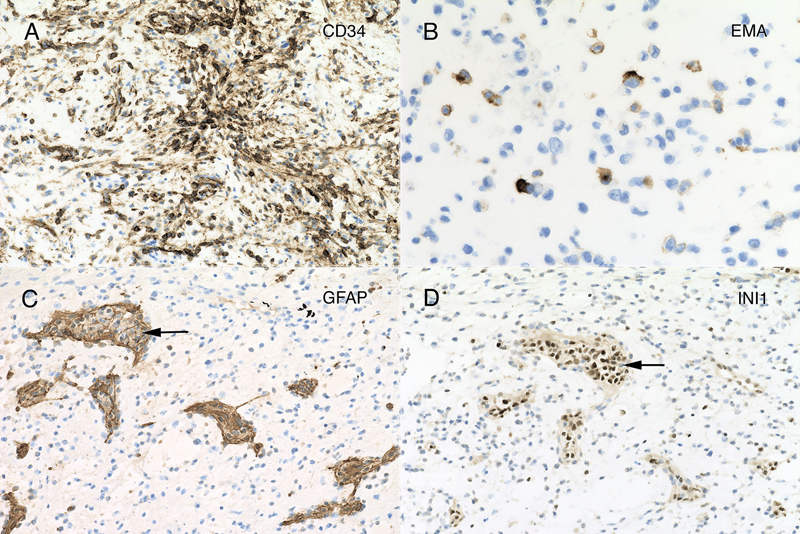
Figure 4: Myxoid matrix and desmoplasia. Matched panels A – B (10x) and C – D (20x) are from the separate histologic regions. The hematoxylin-eosin section in panel A includes several microcysts (black arrow), which stain with Alcian blue (B, black arrow). In addition, the loose, slightly basophilic myxoid matrix in A and C also stains strongly for Alcian blue (B). Throughout the tumor, cells elaborated fine eosinophilic strands (C), which stained strongly blue in the Masson trichrome (D). Clicking the picture will lead you to the full virtual slide (Elastica-van Gieson). Tumor Immunophenotype Immunohistochemistry was performed using the Dako Omnis. The overall results are presented in supplemental table 1. Many areas of the tumor strongly expressed CD34 (figure 5A), which was positive in the perinuclear cytoplasm and to a lesser extent in the long processes extending from the cells. In contrast, only a minor population tumor cells expressed EMA and only in a perinuclear location (panel 5B). However, this EMA expression occurred throughout the tumor. Gliovascular islands, composed of vessels encased within an astrocytic (glial fibrillary acidic protein, GFAP; figure 5C) and synaptophysin-positive matrix (data not shown) were dispersed throughout the tumor. The myxoid component showed no expression of either of these markers, indicating it did not derive from primary brain parenchyma. The most informative stain was INI1, whose expression was lost in the tumor (figure 5D) but retained in the reactive gliovascular islands (black arrow in 5D). These findings indicated that the tumor had 1) a defect in the SMARCB1 gene or its expression and 2) incorporated adjacent brain tissue. While individual tumor cells had cytological features reminiscent of rhabdoid cells, they did not express myogenin or desmin. See supplemental table 1.
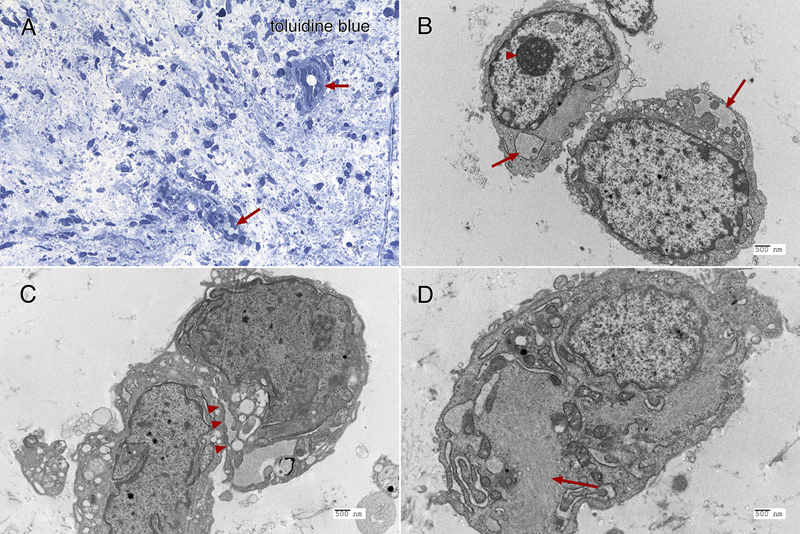
Figure 5: Immunohistochemistry. The tumor strongly immunoreacted for CD34 in many regions (panel A; 20x), in both perinuclear regions and cytoplasmic processes. Although widespread, the tumor showed only perinuclear reactivity for EMA, and only in a small subset of tumor cells (panel B; 40x). Many regions of tumor had islands of neuropil and reactive astrocytes that stained for glial fibrillary acidic protein (GFAP, panel C, black arrow; 20x). These islands retained strong reactivity for INI1 (panel D, from same region as panel C, black arrow; 20x), however, the encasing tumor had lost expression of this molecular marker. Clicking the picture will lead you to the full virtual slide (SMARCB1/INI1). Electron microscopy Toluidine-stained plastic 1-micron sections again showed a relatively hypocellular neoplasm have cells widely disbursed in the loose matrix (figure 6A). Vessels and some surrounding cells represented portions of the gliovascular element (6A, red arrows). Ultrastructurally, tumor cells had scant cytoplasm and large nuclei with prominent nucleoli (figure 6B, red arrowhead). Although by both routine histology and in cytological preparation, the tumor had frequent cells that appeared to have several nuclei, in ultrastructure, at least some of these cells were separated by a plasmalemma (6C, red arrowheads). The ultrastructure of these tumor cells was similar to the cells in the CSF cytology and resected tumor routine stains (compare figure 6 with figures 2 and 3F). The cytoplasm typically contained small vacuoles (6B, red arrows) and intermediate filaments (6D, red arrow).
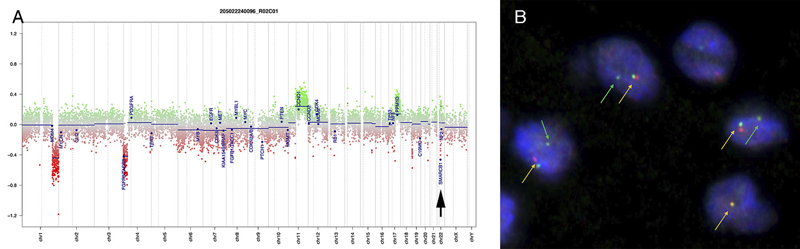
Figure 6: Electron microscopy. The toluidine-stained 1-micron plastic section (A, 40x) demonstrated a hypocellular tumor having cells embedded in a loose matrix, with embedded gliovascular elements (red arrows). In ultrastructural examination using a Hitachi H-7650 electron microscope at 75 kV, the tumor cells have large nuclei with prominent nucleoli (B, arrowhead, and C), scant cytoplasm containing both vacuoles (B, red arrows) and intermediate filaments (D, red arrows). Closely apposed cells were separated by plasmalemma (C, red arrowheads). Molecular Results On DNA-methylation profiling (Illumina Methylation EPIC BeadChip) using the Heidelberg CNS tumor classifier [Capper, 2018], the neoplasm was not classifiable (calibrated score <0.3). Copy number alterations derived from DNA-methylation intensity values (figure 7A) revealed gains of chromosome 11q and 17q, losses of chromosomes 1q and 4p as well as heterozygous loss affecting the SMARCB1 region on chromosome 22q11.23 (black arrow). To confirm the specific loss, SMARCB1 FISH (figure 7B) was performed as described previously [Frühwald, 2020]. This demonstrated a heterozygous loss of SMARCB1 locus.
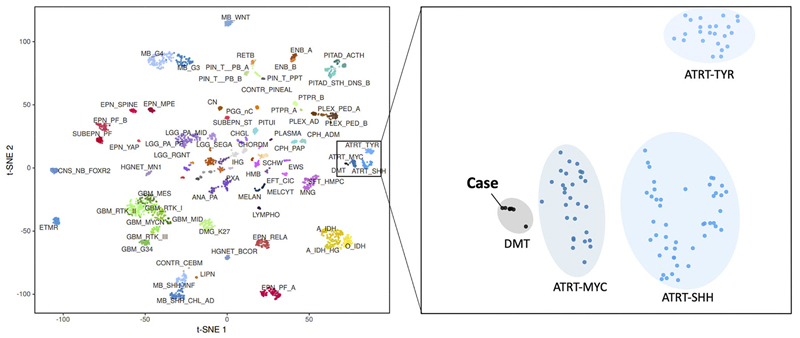
Figure 7: Copy number variation. Copy number alterations (A) derived from DNA-methylation intensity values revealed chromosomal gains (green values) and losses (red values), including losses affecting the SMARCB1 region on chromosome 22q11.23 (arrow). Fluorescence in situ hybridization (FISH) analysis using a probe consisting of the two clones RP11-71G19 (spectrum orange) and RP11-911F12 (spectrum green) (B) showed one colocalized signal per nucleus (orange arrows) representing the intact SMARCB1 locus and a heterozygous loss of the deleted SMARCB1 allele (green arrows) containing the clone RP11-71G19 (loss of red signal) and parts of clone RP11-911F12 (diminished intensity of remaining single green signal). Next, t-distributed stochastic neighbor embedding (t-SNE) analysis was performed in comparison with DNA methylation profiles of 2.801 previously published samples comprising 82 molecularly distinct CNS tumor entities [Capper, 2018] as well as DNA methylation profiles of six previously published DMT cases [Thomas, 2020]. Here, the DNA methylation profile of the present case clearly grouped with that of DMT (figure 8, right, gray oval). This grouping was close to but distinct from three different groupings of SMARCB1-mutant atypical teratoid-rhabdoid tumors (ATRT) (figure 8, right, blue ovals).
Figure 8: DNA methylation studies and SMARCB1 gene analysis. On t-distributed stochastic neighbor embedding (t-SNE) analysis, the DNA methylation profile of 82 distinct tumor entities (left), this patient's tumor grouped with that of DMT (right). Discussion The tumour presented here is histologically and immunophenotypically similar to cases presented in Thomas, 2020, Matsumura, 2021, Wang, 2021. As demonstrated in the DNA methylation studies (figure 8), this tumour also falls into the same cluster as the desmoplastic myxoid tumour, SMARCB1-mutant tumor described in Thomas, 2020. Available demographic data from all prior cases indicate no gender differences (F: 6; M: 4) and an average age of onset of 37 years (range 15-61 years old). Of note, all prior cases have been in the pineal region. The SMARCB1 gene, which is also known as INI1 (Integrase interactor 1) and hSNF5 (SNF5 homolog), encodes the SWI/SNF-related, matrix-associated actin-dependent regulator of chromatin, subfamily B, member 1 protein, which is a core component of the ATP-dependent SWI/SNF chromatin-remodeling complex [Phelan, 1999] and acts as a tumor suppressor [Versteege, 2002]. Biallelic inactivation of SMARCB1 has been associated with the growth of benign and malignant tumors in children and adults [Hollmann, 2011]. Intracranial SMARCB1-deficient tumors span a range of malignancies and include childhood atypical teratoid/rhabdoid tumor (ATRT) [Biegel, 1999], cribriform neuroepithelial tumor [Hasselblatt, 2009], poorly differentiated chordoma [Mobley, 2010], meningeal tumors having challenging histology [Dadone, 2017], a young adult superficial (not pineal) rhabdoid tumour [Bodi, 2018], poorly differentiated sinonasal carcinoma [Agaimy, 2017], and the desmoplastic myxoid tumor, SMARCB1-mutant, of the pineal region (DMT) that we present here [Thomas, 2020; Matsumura, 2021; Wang, 2021]. Two previous reports discussed DMT radiologic features. Unlike the tumour described here, which was hypodense in CT and hypointense in T1-weighted MRI (figure 1A and 1B), the tumor described in Wang, 2021 was hyperdense on CT scans and slightly hyperintense on T1-weighted MRI scans. It is likely that the more prominent desmoplasia illustrated in Wang, 2021, compared to our tumor, correlated with their increased CT signal density. In our patient's tumor, the Alcian blue myxoid component (see figure 5B) was predominant, which correlates with our increased T2 and FLAIR signal (see figure 1C and 1D) and is dissimilar to the isointense T2-weighted signal described in Matsumura, 2021. Similar to the image illustrated in Matsumura, 2021, gadolinium enhancement was heterogeneous in our patient's tumour. This tumor had several features not yet described in DMTs. It had a pervasive myxoid matrix and had little of the overt and possibly reactive desmoplasia illustrated in Thomas, 2020, Wang, 2021, or Matsumura, 2021. It did, however, diffusely elaborate fine, eosinophilic strands similar to those illustrated in Thomas, 2020 and Wang 2021 (see figure 4C and 3D), which stain like collagen (figure 4D). We feel that this represents intrinsic tumor desmoplasia, rather than the reactive desmoplasia illustrated in the other cases and common to many systemic cancers. The DMT also had non-neoplastic elements, including gliovascular islands (figure 5C) that strongly expressed GFAP but retained INI1 expression (figure 5D). These islands likely originated from the underlying pineal gland or surrounding brain. Ultrastructural studies demonstrated the tumour cells had large nuclei with prominent nucleoli. The cytoplasm was scant and had vacuoles and intermediate filaments (figure 6). Unlike the prior reports, our patient had atypical cells in her cerebrospinal fluid (figure 2). Because of the scant available material in the CSF cytology, we have not been able to verify these cells as neoplastic; however, we feel their cytologic features were morphologically similar to those in light (figure 3F) and electron microscopy (figure 6) from the tumor resection and considered them neoplastic rather than chondrocytes. Our tumor cells lacked desmin or myogenin expression. As in our tumor, most cases expressed CD34 (figure 5A). EMA expression has been variable, with strong positivity in 5 of 6 cases in Thomas, 2020, focally positive in Matsumura, 2020, and absent in Wang, 2021. In our patient, the EMA expression was subtle but distinct (figure 5B). Given these data, we feel the salient pathological features of this tumor are:
The advent of integrated genomics has revolutionized our understanding of ATRT through the identification of three molecular subgroups (ATRT-SHH, ATRT-TYR, and ATRT-MYC), which differ in their demographics, location, prognosis, and SMARCB1 mutational profile [Johann, 2016; Torchia, 2016]. Comparative methylation profiling of DMT with other intracranial SMARCB1-mutant tumors has shown DMT, SMARCB1-mutant tumors most closely resemble ATRT-MYC and poorly differentiated chordomas [Thomas, 2020]. By contrast, the superficial CD34+ SMARCB1-deficient tumor described in Bodi, 2018, clustered with the ATRT-MYC, using the Heidelberg Brain Tumor Classifier, and not with the DMT, SMARCB1-mutant group [Thomas, 2020]. The patient we present here also has a methylation profile that clusters together with the other described SMARCB1-mutant myxoid pineal tumours (see figure 8). While it is interesting to speculate about the origins or common features of SMARCB1-mutant tumours, and although it has some features that overlap with ATRT, the neoplasm described here is histologically distinct from other SMARCB1-mutant tumours. References Agaimy A, Hartmann A, Antonescu CR, Chiosea SI, El-Mofty SK, Geddert H, Iro H, Lewis JS Jr, Märkl B, Mills SE, Riener MO, Robertson T, Sandison A, Semrau S, Simpson RH, Stelow E, Westra WH, Bishop JA. SMARCB1 (INI-1)-deficient sinonasal carcinoma: A series of 39 cases expanding the morphologic and clinicopathologic spectrum of a recently described entity. Am J Surg Pathol. 2017 Apr;41(4):458-471. Arya A. Cartilage cells - a potential mimicker of malignant cells in cerebrospinal fluid and a diagnostic pitfall. J Cytol. 2019 Oct-Dec;36(4):218-219. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999 Jan;59(1):74-79. Bigner SH, Jonston WW. The cytopathology of cerebrospinal fluid. I. Nonneoplastic conditions, lymphoma and leukemia. Acta Cytol. 1981 Jul-Aug;25(4):345-353. Bodi I, Giamouriadis A, Sibtain N, Laxton R, King A, Vergani F. Primary intracerebral INI1-deficient rhabdoid tumor with CD34 immunopositivity in a young adult. Surg Neurol Int. 2018 Feb;9(45). Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018 Mar;555(7697):469-474. Chen KT, Moseley D. Cartilage cells in cerebrospinal fluid. Arch Pathol Lab Med. 1990 Feb;114(2):212. Cibas ES. Chapter 6. Cerebrospinal fluid. In Cibas ES, Ducatman BS, editors. Cytology e-book: Diagnostic principles and clinical correlates, fourth edition. 2014, Elsevier, Health Sciences Division, Philadelphia. Dadone B, Fontaine D, Mondot L, Cristofari G, Jouvet A, Godfraind C, Varlet P, Ranchère-Vince D, Coindre JM, Gastaud L, Baudoin C, Peyron AC, Thyss A, Coutts M, Michiels JF, Pedeutour F, Burel-Vandenbos F, RENOP. Meningeal SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily B member 1 (SMARCB1)-deficient tumours: an emerging group of meningeal tumours. Neuropathol Appl Neurobiol. 2017 Aug;43(5):433-449. Frühwald MC, Hasselblatt M, Nemes K, Bens S, Steinbügl M, Johann PD, Kerl K, Hauser P, Quiroga E, Solano-Paez P, Biassoni V, Gil-da-Costa MJ, Perek-Polnik M, van de Wetering M, Sumerauer D, Pears J, Stabell N, Holm S, Hengartner H, Gerber NU, Grotzer M, Boos J, Ebinger M, Tippelt S, Paulus W, Furtwängler R, Hernáiz-Driever P, Reinhard H, Rutkowski S, Schlegel P-G, Schmid I, Kortmann R-D, Timmermann B, Warmuth-Metz M, Kordes U, Gerss J, Nysom K, Schneppenheim R, Siebert R, Kool M, Graf N. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020 Jul;22(7):1006-1017. Hasselblatt M, Oyen F, Gesk S, Kordes U, Wrede B, Bergmann M, Schmid H, Frühwald MC, Schneppenheim R, Siebert R, Paulus W. Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol. 2009 Dec;68(12):1249-1255. Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011 Oct;35(10):e47-e63. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Segura Wang M, Korshunov A, Rhyzova M, Gröbner S, Brabetz S, Chavez L, Bens S, Gröschel S, Kratochwil F, Wittmann A, Sieber L, Geörg C, Wolf S, Beck K, Oyen F, Capper D, van Sluis P, Volckmann R, Koster J, Versteeg R, von Deimling A, Milde T, Witt O, Kulozik AE, Ebinger M, Shalaby T, Grotzer M, Sumerauer D, Zamecnik J, Mora J, Jabado N, Taylor MD, Huang A, Aronica E, Bertoni A, Radlwimmer B, Pietsch T, Schüller U, Schneppenheim R, Northcott PA, Korbel JO, Siebert R, Frühwald MC, Lichter P, Eils R, Gajjar A, Hasselblatt M, Pfister SM, Kool M. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016 Mar;29(3):379-393. Matsumura N, Goda N, Yashige K, Kitagawa M, Yamazaki T, Nobusawa S, Yokoo H. Desmoplastic myxoid tumor, SMARCB1-mutant: a new variant of SMARCB1-deficient tumor of the central nervous system preferentially arising in the pineal region. Virchows Arch. 2021 Jan. doi: 10.1007/s00428-020-02978-3. Epub ahead of print. Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, Hayden MG, Cherry AM, Gokden M, Edwards MS, Fisher PG, Vogel H. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. 2010 Dec;120(6):745-753. Phelan ML , Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999 Feb;3(2):247-253 . Takeda M, King DE, Choi HY, Gomi K, Lang WR. Diagnostic pitfalls in cerebrospinal fluid cytology. Acta Cytol. 1981 May-Jun;25(3):245-250. Thomas C, Wefers A, Bens S, Nemes K, Agaimy A, Oyen F, Vogelgesang S, Rodriguez FJ, Brett FM, McLendon R, Bodi I, Burel-Vandenbos F, Keyvani K, Tippelt S, Poulsen FR, Lipp ES, Giannini C, Reifenberger G, Kuchelmeister K, Pietsch T, Kordes U, Siebert R, Frühwald MC, Johann PD, Sill M, Kool M, von Deimling A, Paulus W, Hasselblatt M. Desmoplastic myxoid tumor, SMARCB1-mutant: clinical, histopathological and molecular characterization of a pineal region tumor encountered in adolescents and adults. Acta Neuropathol. 2020 Feb;139(2):277-286. Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez NR, Lu M, Chan TS, Picard D, de Antonellis P, Khuong-Quang DA, Planello AC, Zeller C, Barsyte-Lovejoy D, Lafay-Cousin L, Letourneau L, Bourgey M, Yu M, Gendoo DMA, Dzamba M, Barszczyk M, Medina T, Riemenschneider AN, Morrissy AS, Ra YS, Ramaswamy V, Remke M, Dunham CP, Yip S, Ng HK, Lu JQ, Mehta V, Albrecht S, Pimentel J, Chan JA, Somers GR, Faria CC, Roque L, Fouladi M, Hoffman LM, Moore AS, Wang Y, Choi SA, Hansford JR, Catchpoole D, Birks DK, Foreman NK, Strother D, Klekner A, Bognár L, Garami M, Hauser P, Hortobágyi T, Wilson B, Hukin J, Carret AS, Van Meter TE, Hwang EI, Gajjar A, Chiou SH, Nakamura H, Toledano H, Fried I, Fults D, Wataya T, Fryer C, Eisenstat DD, Scheinemann K, Fleming AJ, Johnston DL, Michaud J, Zelcer S, Hammond R, Afzal S, Ramsay DA, Sirachainan N, Hongeng S, Larbcharoensub N, Grundy RG, Lulla RR, Fangusaro JR, Druker H, Bartels U, Grant R, Malkin D, McGlade CJ, Nicolaides T, Tihan T, Phillips J, Majewski J, Montpetit A, Bourque G, Bader GD, Reddy AT, Gillespie GY, Warmuth-Metz M, Rutkowski S, Tabori U, Lupien M, Brudno M, Schüller U, Pietsch T, Judkins AR, Hawkins CE, Bouffet E, Kim SK, Dirks PB, Taylor MD, Erdreich-Epstein A, Arrowsmith CH, De Carvalho DD, Rutka JT, Jabado N, Huang A. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 2016 Dec;30(6):891-908. Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002 Sep;21(42):6403-6412. Wang YE, Chen JJ, Wang W, Zhang AL, Zhou W, Wu HB. A case of desmoplastic myxoid tumor, SMARCB1 mutant, in the pineal region. Neuropathology. 2021 Feb;41(1):37-41. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011 Jun;11(7):481-492.
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |