|
|
|
Free Neuropathology 2:13 (2021) |
|
Original Paper |
|
Specific immune modulation of experimental colitis drives enteric alpha-synuclein accumulation and triggers age-related Parkinson-like brain pathology |
|
Stefan Grathwohla, Emmanuel Quansahb, Nazia Maroofa, Jennifer A. Steinerb, Liz Spychera, Fethallah Benmansourc, Gonzalo Duran-Pachecod, Juliane Siebourg-Polsterd, Krisztina Oroszlan-Szovika, Helga Remya, Markus Haenggia, Marc Stawiskia, Matthias Selhausend, Pierre Maliverd, Andreas Wolferte, Thomas Emriche, Zachary Madajb, Arel Sud, Martha L. Escobar Galvisb, Christoph Muellerf, Annika Herrmannd, Patrik Brundinb*, and Markus Britschgia* |
|
a Roche Pharma Research and Early Development, Neuroscience and Rare Diseases Discovery and Translational Area, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, Basel, Switzerland |
|
*Corresponding authors: |
|
Submitted: 07 April 2021 Accepted: 08 May 2021 Copyedited by: Bert M. Verheijen Published: 18 May 2021 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Alpha-synuclein, Experimental colitis, Enteric nervous system, Parkinson’s disease, Substantia nigra |
|
Abstract
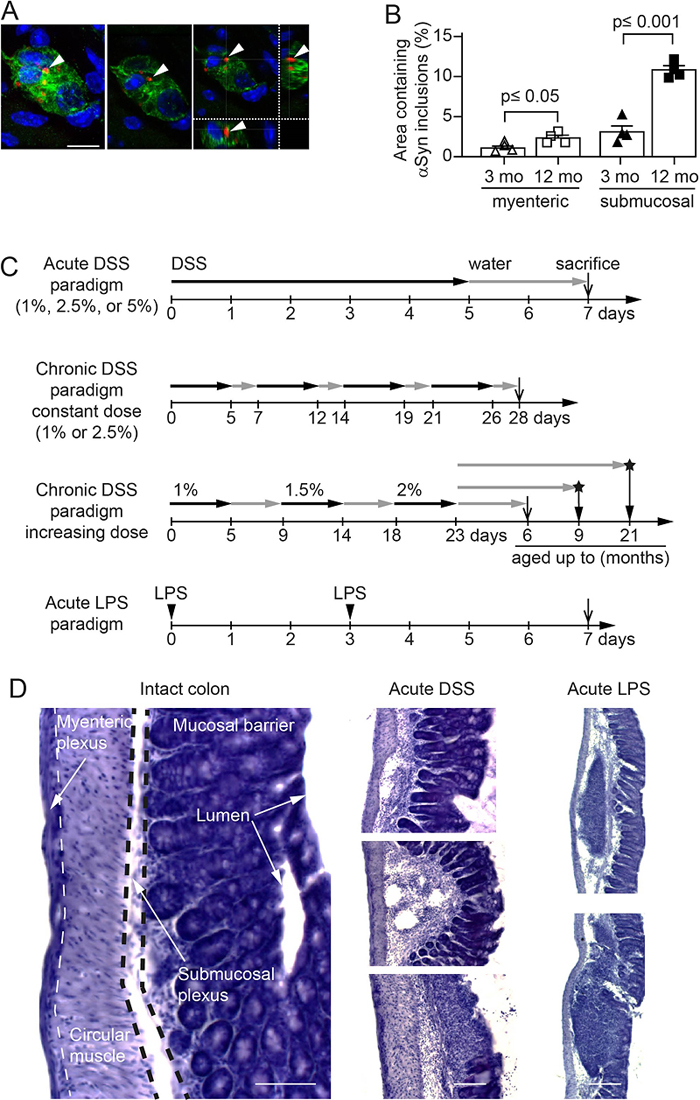
Background: In some people with Parkinson’s disease (PD), α-synuclein (αSyn) accumulation may begin in the enteric nervous system (ENS) decades before development of brain pathology and disease diagnosis. Introduction Parkinson’s disease (PD) is a progressively debilitating neurodegenerative disease affecting 1% of the population above 60 years [1]. Typical symptoms are motor impairments including muscle rigidity, tremor, and bradykinesia. Neuropathologically, PD is hallmarked by loss of dopaminergic neurons in the substantia nigra (SN), a concomitant reduction of striatal dopaminergic signaling [2], and the presence of intraneuronal inclusions called Lewy bodies and neurites [3]. Lewy pathology is enriched in α-synuclein (αSyn), a presynaptic protein that tends to aggregate and become phosphorylated at serine 129 under pathological conditions [2]. Rare point mutations in αSyn and gene multiplications also cause familial forms of PD and related neurological conditions, and certain single nucleotide polymorphisms close to the αSyn gene (SNCA) locus are associated with increased risk for sporadic PD [4]. These findings make αSyn a focal point of biomarker and drug development programs for PD. Several years before the first appearance of motor symptoms, many patients exhibit a variety of non-motor symptoms including constipation, sleep disorder, depression, and hyposmia [5–7]. Indeed, co-occurrence of some of these non-motor symptoms is coupled to elevated PD risk [8–11]. Constipation is an important non-motor feature of prodromal PD, with 28-61% of patients having exhibited gastrointestinal dysfunction for several years during the prodrome [7,10,12]. Notably, αSyn-immunoreactive inclusions have been found in neurons of the submucosal plexus in people with PD [3,13]. Taken together, this converging evidence suggests an early involvement of the enteric nervous system (ENS) in the pathogenesis of PD. Already over a decade ago, Braak and colleagues hypothesized that αSyn-immunoreactive inclusions first appear in the ENS and then occur in the parasympathetic (e.g., vagal output neurons in the intestines) and sympathetic (e.g., in the celiac ganglion in the upper abdomen) nervous system and gradually engage the brainstem, including the vagal dorsal motor nucleus and midbrain areas [3,13]. Several studies in preclinical models have demonstrated that αSyn pathology in the gut is associated with the development of αSyn pathology in the brain [14–21]. For a better understanding of PD pathogenesis and particularly events happening at preclinical stages of PD, it is critical to determine factors that regulate αSyn accumulation in the ENS and to understand whether the process underlying αSyn accumulation in the gut can also lead to αSyn pathology in the brain. Inflammation can potentially trigger αSyn pathology in the ENS of the gut and in the brain. A recent finding in children with gastrointestinal inflammation suggests an immune regulatory function of αSyn [22]. Immune pathways are indeed activated in the brain and colon of PD cases [23,24]. Also, several genes associated with an increased PD risk have an immune system-related function [25], and it was recently proposed that PD heritability is not simply due to variation in brain-specific genes, but that several cell types in different tissues are involved [26]. Further genetic evidence supporting a role for immune pathways in PD pathogenesis is provided by a genome-wide association study that identified common genetic pathways linking PD and autoimmune disorders [27]. Most prominently, LRRK2, a major genetic risk factor for PD [28] also confers increased risk for developing inflammatory bowel disease (IBD) [29]. Certain risk alleles are shared between PD and Crohn’s disease [30], and LRRK2 is known to modulate the function of monocytes, macrophages and other immune cells [31,32]. Intriguingly, IBD is associated with an increased risk for developing PD and specifically blocking the tumor necrosis factor (TNF) pathway reduces this risk [33–37]. Recently, it was reported that experimental colitis in αSyn transgenic mice leads to enteric accumulation of αSyn and the development of PD-like brain pathology and symptoms within a few months [38]. Converging clinical and nonclinical data suggest that the intestinal immune environment plays a role in triggering PD or facilitating the molecular events involved in the earliest phases of the disease process [39,40]. Here, we tested the hypothesis that specific types and severity of intestinal inflammation are required to trigger the accumulation of αSyn in the ENS and the subsequent development of αSyn pathology in the brain. Experimental forms of colitis in wild type and αSyn transgenic mice demonstrated that the type and degree of inflammation regulates the amount of αSyn accumulation in the colon. Macrophage-related signaling limited the extent of αSyn immunoreactivity as demonstrated in a genetic and a pharmacological immune modulation paradigm in the experimental colitis mouse model. Most remarkable, when αSyn transgenic mice were exposed to experimental colitis at 3 months of age and then were allowed to age normally up to 9 or 21 months, the accumulation of aggregated αSyn in midbrain, including the SN, was much exacerbated in the 21-month-old group, but not in the 9-month-old group. These 21-month-old mice also exhibited loss of nigral tyrosine hydroxylase-immunoreactive neurons. Together, our data provide experimental evidence in mice that certain specific forms of intestinal inflammation might be a relevant upstream trigger that plays a critical role in the initiation of PD pathogenesis and the disease progression. Methods Aim, design and setting We aimed to combine an αSyn transgenic mouse model of age-dependent development of αSyn pathology with well-established experimental colitis paradigms in order to explore the effect of type and severity of immune activation on the development of αSyn pathology in the colon and the brain. The design and setting of the different studies are illustrated in Fig. 1.
Fig. 1 Age-dependent increase of intracellular αSyn accumulation in enteric nervous system of hemizygous (Thy1)-h[A30P]αSyn transgenic mice and setup of the experimental colitis paradigms. Mice Male C57BL/6J wild type mice (Jackson Laboratories, Bar Harbor, USA), hemizygous Tg(Thy1-SNCA*A30P)18Pjk ((Thy1)-h[A30P]αSyn) [41] and Tg(Thy1-SNCA*A30P)18Pjk crossed with Cx3cr1tm1Litt ((Thy1)-h[A30P]αSyn /Cx3cr1-def; homozygous for Cx3cr1-GFP knock-in allele; [42] transgenic mice were used for the study. (Thy1)-h[A30P]αSyn transgenic mice express mutant human αSyn under the neuron selective Thy1 promoter. (Thy1)-h[A30P]αSyn transgenic mice were crossed to Cx3cr1-def transgenic mice which express eGFP replacing fractalkine receptor gene expression. All mice were maintained on a C57BL/6J background for more than 10 generations and under specific pathogen-free conditions. To the extent possible, littermates were used in the experiments. Health status was monitored daily during experiments. The in vivo experiments were endorsed by a Roche internal review board and approved by the local animal welfare authorities of the Canton Basel-Stadt, Basel, Switzerland. Experimental colitis paradigms in mice Paradigms for the induction of inflammation were either 1 week (acute) or 3-4 weeks (chronic) with or without an incubation phase under normal conditions of 2-, 6-, or 18-months post application (Fig. 1). Acute systemic inflammation was induced by intraperitoneal (i.p.) lipopolysaccharide (LPS) application [43] of 0.5 mg/kg in 100 µl injection volume on day 0 and 3 (Sigma-Aldrich Chemie GmbH, Steinheim, Germany, LPS 055:B5). Acute colitis was induced by application of 36-50kDa dextran sulfate sodium (DSS) [44] (160110, MP Biomedicals, LLC, Illkirch, France) at 1%, 2.5% or 5% in autoclaved drinking water for 5 continuous days respectively, followed by 2 days of water (1 DSS application cycle). Chronic colitis was induced by two different dosing protocols: i) in a constant dose of DSS (1% or 2.5%) for 5 days and changed to 2 days with normal drinking water and repeated three times (4 application cycles in total); ii) in an increasing dose of DSS starting at 1% for 5 days followed for 4 days on normal drinking water, then increased to 1.5% DSS for the next 5 days followed by 4 days of water and a final cycle of 2% DSS followed by aging the mice on normal drinking water until they were sacrificed. Mice from the same littermate group were randomized per cage into ‘exposed to inflammation inducing agent’ (LPS or DSS, respectively) or ‘unaffected’ (vehicle for the LPS paradigm or normal drinking water for the DSS paradigms, respectively). For the long-term experiments with the two aging cohorts ‘9 months’ and ‘21 months’, respectively, all mice in that study were simultaneously exposed in one large cohort at the age of about 3 months to the increasing dose chronic DSS paradigm (Fig. 1) and DSS exposure was stopped for all mice on the same day after the 23-day period. The mice were then kept and aged on normal drinking water and under normal housing conditions in the same room until the day they were perfused and tissue was collected. IL-10 treatment and exposure measurement Two different forms of mouse IgG bound murine IL-10 (mIgG(v1)-mIL10 and mIgG(v2)-mIL10) were diluted in pre-prepared sterile formulation buffer comprised of 0.5% mouse serum supplemented with 25 mM citrate, 300 mM arginine to a final concentration of 0.75 mg/ml and the pH adjusted to 6.7 on the day of application. Each mouse was treated once with 150 µg i.p. concurrently with the initiation of the acute colitis paradigm with 5% DSS. The concentrations of mIgG-mIL10 fusion proteins in murine serum samples were determined by enzyme-linked immunosorbent assays (ELISA) specific for the Fab moiety of the administered mIgG-mIL10 fusion protein. Biotinylated mIgG-mIL10-specific target molecules were used for capturing, goat anti-mIg IgG-HRP conjugate and peroxidase substrate ABTS were used for quantitative detection of mIgG-mIL10 fusion proteins. Immunohistochemistry Mice were injected with a lethal dose of pentobarbital (150 mg/kg). Upon full anesthesia, mice received transcardial perfusion with room temperature phosphate buffered saline (PBS). For biochemical and immunohistochemical analysis, one section of the proximal colon was either fresh frozen and stored at -80°C or post-fixed in 4% paraformaldehyde (PFA) solution for 24 h. Following post-fixation, organs were incubated in 30% sucrose/PBS at 4°C for at least 48 h before further processing. Subsequently, enteric tissue was cryotome-sectioned to 35 µm thick longitudinal sections (approx. 1 cm length). The brain was collected and post-fixed for 24 h in 4% PFA followed by 30% sucrose in phosphate buffer until cryo-sectioning of floating sections at 40 μm. Histological analysis of mouse colon was performed using standard hematoxylin staining. Immunohistochemical staining was accomplished using the Vectastain Elite ABC Kits and Peroxidase Substrate Kit SK-4100 (Vector Laboratories, Burlingame, CA, USA) or fluorescently labelled secondary antibodies (Alexa 488, 555 or 647, Life Technologies, Zug, Switzerland). The following primary antibodies have been used for overnight incubation at a dilution of 1:1000; monoclonal antibody to human αSyn (clone 211, sc-12767, Santa Cruz Biotechnology, Heidelberg, Germany; specific to human αSyn and binds to normal αSyn as well as abnormal αSyn inclusions which contain the respective epitope), monoclonal antibody generated towards rat αSyn, cross-reactive with murine and human αSyn (Syn1/clone 42, BD Transduction Laboratories, Allschwil, Switzerland; used for wild type mice), polyclonal antibody to the peripheral neuronal marker Peripherin (Millipore Corporation, Billerica, MA, USA), and polyclonal antibody to macrophage marker Iba-1 (Wako Chemical GmbH, Neuss, Germany). To detect αSyn phosphorylated at Serine 129 (pSer129-positive inclusions of pathological/abnormal αSyn) in the free-floating brain sections, monoclonal antibody (ab51253, Abcam, Cambridge, USA) was used at a dilution of 1:10000. Prior to the pSer129 staining, the free-floating brain sections were incubated for 10 min at room temperature in a phosphate buffered saline solution containing 10 μg/mL proteinase K (Cat # 25530015; Invitrogen, California, USA). Tyrosine hydroxylase (TH)-immunoreactive cells were detected using a polyclonal antibody (657012, Millipore Sigma) at a dilution of 1:1000. To measure the density of Nissl-positive cells, the TH-stained cells were counter-stained with Cresyl violet. The slides were incubated in 0.1% Cresyl violet solution for 9 min and then dehydrated in 95% and 100% ethanol and then xylene prior to cover slipping with Cytoseal 60 mounting media (Thermo Fisher Scientific). Quantifications of the blind-coded TH/Nissl-stained slides were done using Stereo Investigator (version 2017.01.1; MBF Bioscience, Williams, VT, USA) on an Imager M2 microscope (ZEISS) coupled to a computer. We analyzed 5-7 nigral sections per animal, and a total of 7-8 animals per treatment group. We outlined the substantia nigra pars compacta and counted every TH-immunoreactive and Nissl-positive cell in that area (using a counting frame of 40 µm x 40 µm, grid size of 140 µm x 140 µm, a guard zone of 2 µm and optical dissector height of 20 µm) and then computed the number of cells per section, generating the average cell count per animal. We then calculated the average count of cells per treatment group and analyzed the data using unpaired Student’s t-test after confirming normality and homoscedasticity in Prism 7.0 (GraphPad Software). Imaging and stereological quantification of αSyn deposits in enteric nervous system Imaging and stereological quantification were performed on a Zeiss Axio Imager Z2 fluorescence microscope (Carl Zeiss AG, Jena, Germany). Leica TCS SP5 confocal system using an HCX PL APO CS 40x 1.3 oil UV or an HCX PL APO LB 63x 1.4 oil UV objective was utilized for image recording. Accumulation of αSyn in the ENS (i.e., punctate intracellular bodies/features) was assessed on a random set of 3 adjacent 35 µm thick, αSyn-immunostained sections comprising the myenteric and submucosal neuronal plexuses. Analysis was performed with the aid of a stereology software (Stereo Investigator 10, MBF Bioscience, Williams, VT, USA) as described previously [45]. In the myenteric plexus ganglion, volume was defined by multiple outlined plexuses containing a range of 5-20 neuronal cells and quantified by the optical fraction fractionator technique. In contrast to the myenteric plexus, the submucosa consists of compact plexuses with 1-5 cells including interconnecting neurites. Therefore, the entire submucosa was set as region of interest, analyzed with the area fraction fractionator technique. Results of the submucosal plexus are displayed by percent area containing αSyn deposits. For the IL-10 experiment, αSyn positive inclusions from immunofluorescence images were counted for each image. Inclusion body-like features were filtered based on having a size between 12 and 50000 pixels and a minimal intensity value greater than 300. The filtering step was performed to exclude small background features and macrophages (very large spots). The counts were then aggregated to the animal level by summing the inclusion feature counts of all images per animal and then normalizing for (i.e., dividing by) the number of images for a given animal. Upon exploratory data analysis two mice were excluded: one mouse because it only had one image (technical outlier, missing data point; repeating the staining for this one mouse would have required re-staining the entire cohort in order to be consistent with staining conditions for quantification; this was unnecessary after statistical analysis) and another due to it being an outlier, based on its infiltration score and image data (i.e., in contrast to the other mice that had received DSS, this mouse did not show signs of inflammation or colonic tissue damage that is normally induced by DSS; it could not be determined if that mouse was correctly dosed with DSS and thus it was excluded from the analysis). Quantification of leukocytes infiltration To determine the leukocyte covered area in the colon after LPS or DSS application, three adjacent hematoxylin-stained sections were quantified. Total area of colon sections and localizations of leukocyte assemblies within the tissue architecture were identified and outlined utilizing a stereology software (Stereo Investigator 6, MBF Bioscience, Williams, VT, USA). Percentage of leukocyte covered area has been set in proportion to total area of the analyzed colon section, e.g., to at least the length of 1 cm of proximal colon. For the IL-10 experiment, hematoxylin-stained colon slices were examined by an expert pathologist blinded to treatment conditions. A score of 0-3 was assigned to each section for each of the 3 layers lamina propria, submucosa and muscularis based on the degree of inflammatory infiltration. A score of 0 denoted no inflammation and a score of 3 indicated extensive infiltration. The mean of the values for all 3 layers was taken as the final measure of leukocyte infiltration per mouse. Quantification of Iba-1/αSyn-double positive macrophages The number of Iba-1/αSyn-double positive cells was evaluated by quantification of 10 random regions in 2 adjacent sections of the proximal colon. The region of interest was set to contain the myenteric plexus/circular muscle layer and the submucosal plexus. Scoring of pSer129 pathology and brain heatmap
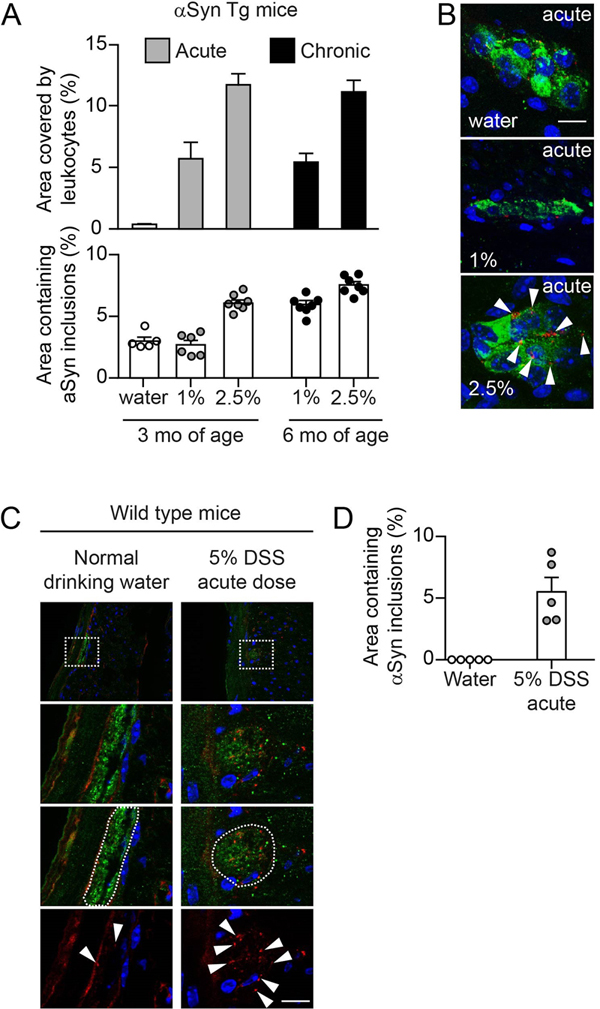
We evaluated pSer129 pathology on a full series of immunostained coronal sections from 10 mice per treatment group (i.e., water vs. DSS-treated groups) on blind-coded slides using a previously described method [46]. We visualized pathology from one hemisphere of all brain sections (apart from the olfactory area) using a NIKON Eclipse Ni-U microscope and assigned scores ranging from 0 to 4 to each brain area based on the relative abundance of proteinase K (PK)-resistant pSer129-positive inclusions (i.e., cell bodies and neurites). In this case, 0 = no aggregates, 1 = sparse, 2 = mild, 3= dense, 4 = very dense. For the heatmap, we obtained the average score values of each brain area for each treatment group. The average data for each treatment group (n=10 mice/group) was then represented as a heatmap in a sagittal mouse brain background. To create the brain heatmap a postscript file downloaded freely from Allen Brain Atlas (mouse, p56, sagittal, image 15 of 21; -> weblink*) was converted to an XML in R v 3.4.4, and the mean scores were manually assigned to respective brain regions. The remaining brain regions were estimated via the R package ‘Akima’, using a pointwise bivariate interpolation algorithm for irregular data on the mean X and Y coordinates for each brain region. Densitometry of pSer129 αSyn brain pathology The density of pSer129 pathology in 12 major brain areas (reticular nucleus, pontine reticular nucleus, periaqueductal gray, gray and white layer, reticular formation, substantia nigra, ventral tegmental area, thalamus, hypothalamus, central amygdala, pallidum and striatum) was determined in the water and DSS-treated animals. A NIKON Eclipse Ni-U microscope was used to acquire 20x magnification images (without condenser lens) from all the indicated brain areas, using the same exposure time for all images. In all cases, images were acquired on three sections separated by 420 μm intervals (localized between Bregma). We then processed the acquired images using Image J64 [47], created a mask (to exclude background) that redirects to the original image for analysis, measured the total area and the mean grey value of the area that had inclusions. For brain areas such as periaqueductal gray that do not fill the entirety of the field to be analyzed, we drew a contour of the area and the analysis was performed only within that contoured area. We subsequently calculated the grey value of the area per square pixels for each image (i.e., A.U./px2 = mean grey value x area stained/total area assessed). Based on this, we calculated the average grey value per square pixels for each brain area for each animal (n = 6 mice/group), and then extended this calculation to determine the average grey value per square pixels for each treatment group and each of the twelve brain areas of interest. Blinding of experimenters for histological and immunohistochemical analyses For analyses of colon and brain tissue on slides, a second individual assigned unique codes to stained slides. Therefore, the experimenter conducted the analyses blinded to the identity of the mice. For randomization of treatment groups see above. mRNA expression To assess mRNA expression levels from the proximal colon, RNA was extracted from fresh frozen tissue with MagnaLyser green beads (Roche Diagnostics, Mannheim, Germany) and Qiazol Lysis (Reagent cat.no.79306, Hilden, Germany) purified on MagnaPure LC (HP Kit no.03542394001, F. Hoffmann-La Roche AG, Rotkreuz, Switzerland) and amplified via real-time PCR (4 ng RNA/reaction; Lightcycler 480, Roche Diagnostics Corporation, Indianapolis, USA). Amplification of mRNA was performed by using TaqMan probes for human or murine specific α-synuclein and for selected cytokines/chemokines (Applied Biosystems Europe B.V., Zug, Switzerland). Target mRNA was normalized to tissue specific murine GAPDH levels and displayed as relative expression after 30 amplification cycles. Statistics Measurements for inflammation and αSyn accumulation in the ENS were taken from distinct samples (e.g., in three to six technical replicates per mouse). Data from each mouse was used only once, thus no repeated measure of the same sample was performed. Statistical analysis of gut pathology and inflammation was performed using GraphPad Prism 6.04 or 7.0 software (GraphPad Software, Inc. La Jolla, CA, USA). The results are expressed as mean values ± standard errors of the mean (S.E.M.). Student’s t-test (or Welch’s t-test for unequal variances) was used to compare two groups and ANOVA was used for multi-comparison of groups followed by Tukey HSD post-hoc analysis. For the statistical analysis of the mRNA expression, data quality was assessed by inspecting the distribution of Cp values of reference endogenous genes across samples, by inspecting the level of Cp variation between technical replicates and by exploring the samples multivariate signal distribution as in a principal component analysis. Relative gene expression levels were expressed as 2-(Cpgene – CpRef). Statistical analyses to assess the effect of the experimental conditions on the log2 gene expression levels were done with linear models using the limma package (Bioconductor/R, [48]). These analyses were implemented in R v 3.1.1. For the statistical modelling of the effects of the IL-10 treatment on αSyn counts, as well as infiltration scores, the levels for IgG1(v1)-IL10 and IgG1(v2)-IL10 treatment were compared to the positive (vehicle/DSS) control. Additionally, since levels of the control antibody treatment (IgG1(v1)) were very similar to the positive control, the two groups were pooled in further contrasts in which effects of individual antibodies or control IgG was assessed. For αSyn counts in the enteric nervous system, a linear model on the treatment groups with one-degree freedom contrasts was applied. For the infiltration score a Kruskal-Wallis test, with the same contrasts, was used. All statistical tests were two-tailed with a significance level of p <0.05. For the statistical analysis of the pSer129 αSyn brain pathology, zero-inflated negative-binomial mixed-effects models with a random intercept for each sample and variance assumed to increase linearly with the mean (verified against a quadratic increase using Akaike Information Criterion [AIC] and Bayesian Information Criterion [BIC]) were used to analyze the dataset via the ‘glmmTMB’ package in R v 3.4.4. Linear contrasts with false discovery rate (FDR) adjustments were then used to test our hypotheses and account for multiple testing (for brain area and experimental group). Results Experimental colitis exacerbates αSyn load in the submucosal plexus of αSyn transgenic and wild type mice During the process of further characterizing a (Thy1)-h[A30P]αSyn transgenic mouse line [41], we detected by immunohistochemistry human αSyn accumulation in all innervated organs that were analyzed (Suppl. Fig. 1). To examine the localization of αSyn inclusions in nervous structures in the ENS, we performed an immunofluorescent co-staining for human αSyn (clone 211) and peripherin, a specific marker for peripheral nerves. By applying confocal microscopy, we established a process and protocol to identify myenteric and submucosal plexuses in order to quantify the αSyn inclusions found in the nerve cells of the ganglia in the colon. The intracellular presence of the irregularly sized and shaped αSyn inclusion bodies was confirmed in 2D z-stacks of rotated confocal images (Fig. 1A). We observed an age-dependent (mice aged 3 months versus 12 months) increase of baseline human αSyn inclusions in both plexuses (Fig. 1B). Given the clinical and epidemiological link between IBD and PD, we wanted to test whether different types and strengths of IBD-related experimental inflammation in the colon exacerbates this local accumulation of αSyn acutely (e.g., within a few days or weeks) and how the age of the αSyn transgenic mice may influence the outcome. Administration of dextran sulfate sodium (DSS) in the drinking water in acute or chronic dosing paradigms are well-established mouse models of experimental colitis mimicking aspects of IBD, i.e., by exhibiting infiltration of leukocytes into the submucosa with various degrees of destruction of the colonic mucosa and submucosa [49]. It is well-known that the effects induced by the DSS paradigm can vary substantially based on the genetic background of the mice and due to different animal housing environments. Thus, in order to establish the DSS paradigm in our environment and with our mice, we first tested the effect of DSS administration at different doses and durations in the (Thy1)-h[A30P]αSyn transgenic mice (Fig. 1C). We observed that leukocyte infiltration was appropriately modulated by the acute dose (1% or 2.5% DSS for 5 days followed by 2 days drinking water) and chronic constant dose (1% or 2.5% DSS alternating with normal drinking water for 28 days, respectively) DSS paradigms (Fig. 1D and 2A). In the same experiment, we wanted to test for an age effect on a potential aggravation of αSyn accumulation in the ENS and applied the acute dose paradigm on 3-month-old mice and the chronic dose paradigm was started at the age of 5 months leading to a final age of 6 months at analysis of the mice. In the acute DSS dosing paradigm with mice at the age of 3 months, 2.5%, but not 1%, DSS triggered intracellular accumulation of αSyn in peripherin positive nerve cells of the submucosal plexus (Fig. 2A, B). In the 28-day chronic constant DSS dose paradigm in mice aged 6 months (Fig. 1C and 2A), we observed that the 1% constant chronic DSS dose showed a similar degree of αSyn inclusions as the 3-month-old mice which were on an acute 2.5% DSS dose paradigm. In addition, mice previously exposed to 2.5% constant chronic DSS dose presented on average with a slightly but robust increased percent area of αSyn inclusions compared with the 6-month-old mice that were on 1% DSS (Fig. 2A). Together, this demonstrated that different DSS paradigms can be established in (Thy1)-h[A30P]αSyn transgenic mice and that different DSS paradigms can induce robust elevation of αSyn inclusions. This first experiment also provided us with data points to estimate a potential effect size for triggering αSyn inclusions in the different DSS paradigms. We observed that the younger (Thy1)-h[A30P]αSyn transgenic mice showed a robust increase in αSyn inclusions and better signal-to-noise conditions of the autofluorescent colonic tissue than the older ones, experiments were henceforth continued with mice of the younger age. In this initial experiment, it was also interesting to observe that (Thy1)-h[A30P]αSyn transgenic mice exposed to acute 2.5% DSS colitis presented with several αSyn-positive cells with a morphology consistent with them being infiltrating leucocytes, which was confirmed by an Iba-1 co-staining (Suppl. Fig. 2). This finding was relevant for the quantification of αSyn inclusions in the myenteric and submucosal plexus, i.e., such features were excluded from the quantification process.
Fig. 2 Experimental DSS colitis severity and duration-dependent aggravation of accumulation of αSyn inclusions in the colonic submucosal plexus of hemizygous (Thy1)-h[A30P]αSyn transgenic and wild type mice. Wild type mice also express endogenous αSyn in innervated organs, but at much lower levels compared with the levels of human αSyn expressed by the hemizygous (Thy1)-h[A30P]αSyn transgenic mice (Suppl. Fig. 1). To confirm that the finding in (Thy1)-h[A30P]αSyn transgenic mice was independent of transgenic expression of human αSyn, we applied an acute 5% DSS dose paradigm (5 days DSS + 2 days water) in wild type mice (Fig. 1C). We observed, in the submucosal plexus, small inclusion bodies of endogenous murine αSyn (detected by rodent cross-reactive αSyn-specific monoclonal antibody Syn1/clone 42, Fig. 2C, D). These features were close to undetectable in the water group that did not experience experimental colitis. A separate experiment also confirmed that the observed effects of elevated αSyn inclusions following acute DSS (5% dose) could not be attributed to increased gene expression of murine or the transgenic human αSyn (Suppl. Fig. 3). Together, these results confirmed the validity of this experimental IBD paradigm to test the effect of inflammation on αSyn accumulation in the ENS in wild type and (Thy1)-h[A30P]αSyn transgenic mice. Because the (Thy1)-h[A30P]αSyn transgenic mouse model is well-established to analyze human αSyn-related pathology, we focused for the remainder of the study on employing these transgenic mice. Colitis induced by peroral DSS but not by intraperitoneal administration of LPS aggravates αSyn accumulation in colonic submucosal plexus of αSyn transgenic mice Different inflammatory agents induce different types of immune stimulation and thus can influence the phenotype of experimental colitis. A well-established experimental immune trigger is the bacterial endotoxin LPS. In order to explore the effects of different approaches to induce inflammation in or nearby the gut in (Thy1)-h[A30P]αSyn transgenic mice, we compared the outcome of acute (5 days DSS + 2 days normal drinking water) 5% DSS per os with acute 0.5 mg/kg intraperitoneal LPS administration (Fig. 1C and 3). To maximize the inflammatory response, we administered both DSS and LPS at relatively high doses. At day 7, both agents had induced variable degrees of leukocyte infiltration in the submucosa of the colon while a marked destruction of the mucosa was induced when giving only DSS (Fig. 1D). As before, the DSS-exposed mice presented with increased accumulation of αSyn in the ganglia of the submucosal plexus (Fig. 3A). In contrast, we detected no change in αSyn load in the myenteric plexus, consistent with lack of leukocyte infiltration in this part of the colonic wall (Fig. 3B). Despite the high dose, LPS-induced inflammation did not increase αSyn accumulation in the colonic nervous plexuses (Fig. 3C, D). Notably, LPS and DSS resulted in a differential expression of cytokines, and consistent with leukocyte recruitment, CCL2 was elevated in both (Fig. 3F, G). In the LPS paradigm, mRNA for IL-10 was markedly elevated, whereas DSS strongly increased IL-6 and also IL-1β but not IL-10. Together these results indicate that, in our model, colonic inflammation induced by peroral DSS but not intraperitoneal LPS increases the accumulation of αSyn in the colon.
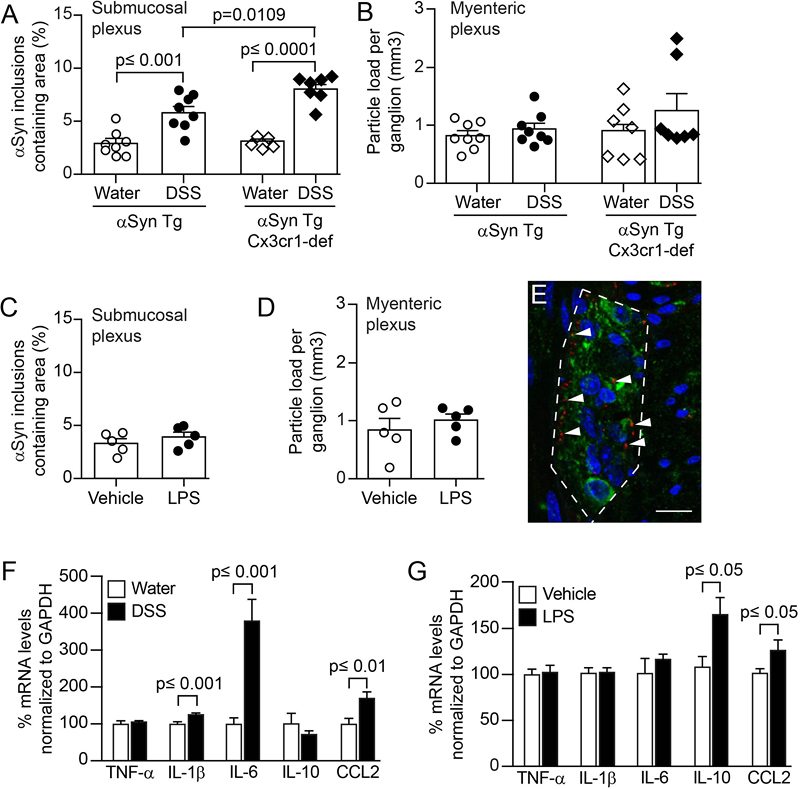
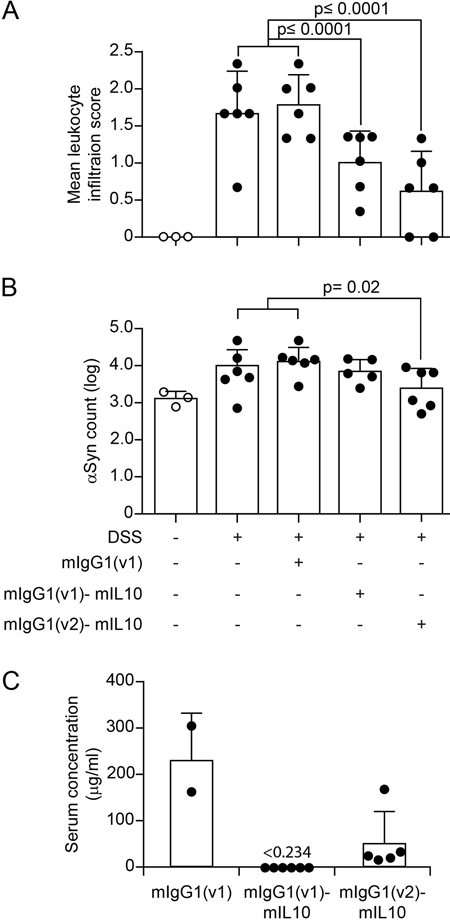
Fig. 3 Colitis induced by peroral DSS but not peritoneal LPS enhances αSyn accumulation in the colonic submucosal plexus of hemizygous (Thy1)-h[A30P]αSyn transgenic mice and can be increased by lack of monocyte/macrophage-related Cx3cr1 signaling. Lack of monocyte/macrophage related Cx3cr1 signaling during DSS colitis increases αSyn load in the submucosal plexus of αSyn transgenic mice Given the role of monocytes/macrophages in IBD and in the related DSS paradigm, we hypothesized further that modulating monocytes/macrophages may affect accumulation of αSyn in our DSS model as well. In a first set of experiments we manipulated monocytes/macrophages genetically by crossing (Thy1)-h[A30P]αSyn transgenic mice with mice that have a deletion for the fractalkine receptor Cx3cr1 (Cx3cr1-GFP knock-in mice) (Fig. 3A, B). The CX3CR1-CX3CL1 axis plays an important role in maintaining the function of the lamina propria macrophage population of the gastrointestinal wall and lack of this signaling pathway in experimental colitis models may either aggravate or ameliorate the induced pathology [50–52]. In our experiment, the area covered by infiltrating leukocytes following exposure to DSS was near the mucosa and submucosa and was not significantly higher in the Cx3cr1-deficient αSyn transgenic mice than in the Cx3cr1-competent mice (Suppl. Fig. 3A). However, a significantly higher level of αSyn accumulated in the submucosal plexus in αSyn transgenic mice lacking Cx3cr1 compared to αSyn transgenic mice expressing Cx3cr1 (p = 0.001, two-way ANOVA with Tukey HSD post-hoc analysis; Fig. 3A). In the myenteric plexus, we found no marked increase in αSyn accumulation in neither the αSyn transgenic mice with normal Cx3cr1 nor the αSyn transgenic mice deficient in Cx3cr1, indicating as in the experiments above a possible prominent role for the localization of leukocyte infiltration in the process of αSyn accumulation in the submucosa (Fig. 3B). Collectively, our results in Cx3cr1-deficient αSyn transgenic mice provide a potential association between monocyte/macrophage signaling and αSyn accumulation in ENS in this experimental IBD model. Systemic IL-10 reduces DSS-induced colitis and associated enteric αSyn accumulation in αSyn transgenic mice To continue testing the hypothesis that modulating monocytes/macrophages may affect accumulation of αSyn in our DSS model we moved to a pharmacological modulation of this cellular subset. Interleukin-10 (IL-10) is an important regulator of monocytes/macrophages, and genetic ablation of IL-10 signaling or blocking IL-10 with specific antibodies has been reported to enhance DSS colitis [53,54]. In the experiments with LPS we had also noted an increase of IL-10 compared with the DSS paradigm and LPS inflammation was in contrast to DSS colitis not associated with increased αSyn accumulation in the ENS (Fig. 3). To mimic the effect of higher levels of IL-10 in an acute model of DSS colitis (5% DSS for 5 days + 2 days normal drinking water, Fig. 1C), we administered intraperitoneally recombinant murine IL-10 (mIL10) in this paradigm. The half-life of injected recombinant IL-10 protein in blood is very short. To reduce the number of injections, we extended the half-life of mIL-10 in circulation by engineering it onto two different murine IgG variants (i.e., mIgG1(v1)-mIL10 and mIgG1(v2)-mIL10, respectively). As described above, DSS induced a marked increase in leukocyte infiltration and αSyn accumulation, and we found both to be similar in the untreated and control IgG treated group (Fig. 4A, B). In contrast, both mIgG1(v1)-mIL10 and mIgG1(v2)-mIL10 significantly reduced leukocyte infiltration in mice treated with DSS (p<0.0001, one-way ANOVA with Tukey HSD post-hoc analysis; Fig. 4A, B). A significant down-regulatory effect of an IL-10 treatment on DSS colitis induced accumulation of human αSyn in the submucosal plexus was only observed with mIgG1(v2)-mIL10 (p=0.02, one-way ANOVA with Tukey HSD post-hoc analysis; Fig. 4B). This effect by mIgG1(v2)-mIL10 on αSyn levels was accompanied by detectable serum levels of mIgG1(v2)-mIL10 at the end of the in vivo phase, whereas mIgG1(v1)-mIL10 was no longer detectable at that point (Fig. 4C). This indicates that although both forms of IL-10 have a down-regulatory effect on leukocyte infiltration, a sustained pharmacological exposure of IL-10 may be required for reducing αSyn accumulation. These results highlight an important role for the IL-10 pathway in keeping αSyn accumulation at a reduced level throughout the course of experimental IBD. Together, our observations by genetic (i.e., CX3CR1-CX3CL1 axis) and pharmacological modulation (i.e., IL-10) of DSS colitis corroborate an important role for monocyte/macrophage pathways in the development of αSyn accumulation in the ENS of the colon.
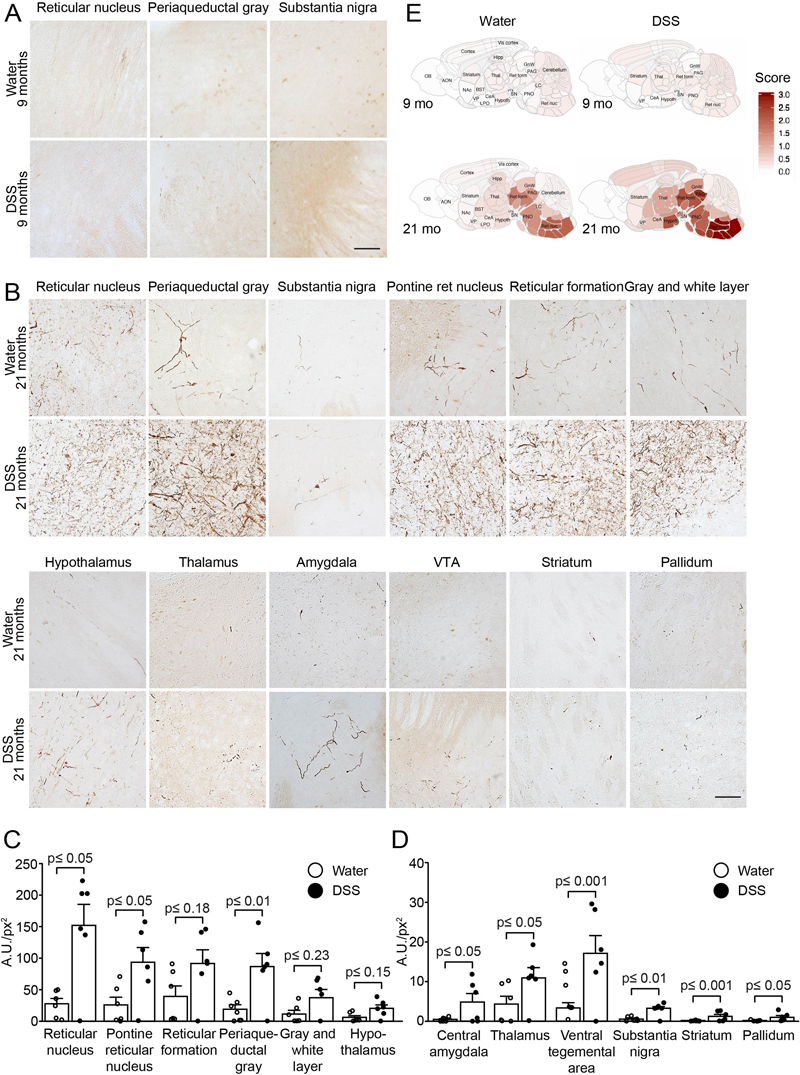
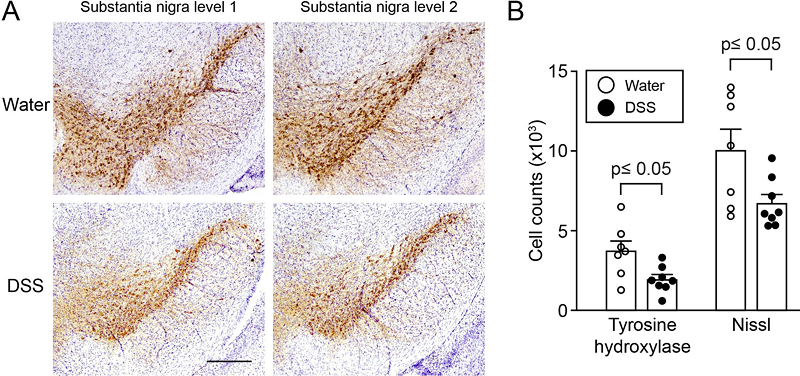
Fig. 4 Systemic IL-10 ameliorates DSS colitis and slightly reduces associated local αSyn accumulation in (Thy1)-h[A30P]αSyn transgenic mice. Two different recombinantly engineered and murine IgG1-fused forms of murine IL-10 (mIgG1(v1)-mIL10 and mIgG1(v2)-mIL10) were administered (150 µg per mouse i.p.) at the beginning of the acute DSS paradigm (5%) in (Thy1)-h[A30P]αSyn transgenic mice. Vehicle and the mIgG1(v1) alone served as untreated controls. A Leukocyte infiltration was assessed by visual scoring and (B) inclusion features of αSyn were stereologically and semi-automatically quantified and result log scaled for statistical analysis. Both the vehicle group and the mIgG1(v1) group had similar levels of leukocyte infiltration and αSyn inclusions and were merged for the statistical analysis to compare with the IL-10 treated groups. Both forms of IL-10 ameliorated leukocyte infiltration whereas mIgG1(v2)-mIL10 also blocked the appearance of αSyn inclusions significantly (n = 3-6 per group; mean and S.E.M.; one-way ANOVA and Tukey post hoc test). C Persistent exposure mIgG1(v2)-mIL10 versus mIgG1(v1)-mIL10 (lower limit of detection is indicated at <0.234 μg/ml) as measured in serum at the end of the in vivo phase corresponds with beneficial treatment effects on αSyn readout observed above. The mIgG1(v1) was only measured in two mice. DSS colitis-induced submucosal αSyn accumulation at a young age persists for months and is exacerbated by lack of Cx3cr1 signaling In humans there is strong epidemiological evidence that IBD increases PD risk [33,35,37] and recent evidence in Crohn’s disease [55] indicates that such gut inflammatory conditions are associated with αSyn accumulation in the ENS [36]. In these experiments in mice, we have until here established and replicated, in different setups, a link between modulation of inflammation and induction of αSyn accumulation in the ENS. Because longer exposure to DSS (i.e., over several weeks) mimics more closely the chronic nature of IBD, we elected to explore αSyn accumulation in the submucosal plexus of (Thy1)-h[A30P]αSyn transgenic mice that were subjected to DSS colitis in a 4-week chronic increasing dose paradigm. In addition, the dose increase with longer water intervals is more gentle for the mice from an animal welfare perspective. In order to allow for a full recovery from the chronic inflammation, we aged the mice for two months on normal drinking water and analyzed them at the age of 6 months (Fig. 1C). At this point we wanted again to explore the effect of modulating monocytes/macrophages in this chronic setting and added an experimental arm with (Thy1)-h[A30P]αSyn transgenic mice lacking Cx3cr1. As expected, after 2 months of recovery, the area that is usually extensively covered by leukocytes in the submucosal plexus of the acute DSS paradigm had returned to normal levels following the two-month recovery period (Suppl. Fig. 4A). Remarkably, however, the area containing αSyn inclusions in the ganglia of the submucosal plexus was still almost doubled when compared to αSyn transgenic mice that were not exposed to DSS, and this was exacerbated in αSyn transgenic mice deficient for Cx3cr1 (Suppl. Fig. 4B). The finding in the αSyn transgenic mice suggests that accumulation of αSyn is not a transient effect or response. In addition, modulation of monocytes/macrophages by down-regulating the CX3CR1-CX3CL1 axis contributes to aggravation of this accumulation. Experimental DSS colitis-induced at a young age exacerbates αSyn brain pathology and dopaminergic neuron loss in old αSyn transgenic mice At this point, we have established and repeatedly demonstrated that modulation of inflammatory mechanisms in experimental colitis induced by acute and chronic DSS administration is causatively linked to induction and persistence of intracellular αSyn inclusions in the ENS of young adult mice. The previously highlighted hypothesis by Braak and colleagues associates αSyn brain pathology in PD with αSyn pathology in the ENS earlier in life [3,56]. To assess development of brain αSyn pathology and to link it to IBD risk, we exposed 3-month-old hemizygous (Thy1)-h[A30P]αSyn transgenic mice to a chronic increasing dose DSS paradigm or normal drinking water and after 23 days returned all mice to normal drinking water until sacrifice several months later (Fig. 1C). We chose to use the αSyn transgenic model rather than wild type mice for this study because of two reasons: 1) we knew that the model as hemizygous transgenic mice exhibit some αSyn brain pathology that develops slowly under baseline conditions. Importantly, the pathology is much less pronounced than in homozygous (Thy1)-h[A30P]αSyn mice [57]; 2) at the time of the experiment, it was not clear whether wild type mice could develop αSyn brain pathology upon DSS colitis. Thus, we chose hemizygous (Thy1)-h[A30P]αSyn transgenic mice to increase the chances for a successful outcome and potentially to aggravate the brain pathology from mild to strong. After exposing the mice either to normal drinking water or a chronic increasing dose DSS paradigm, we aged them in two cohorts on normal water and housing conditions to either up to the age of 9 months or 21 months. At these two timepoints we analyzed various brain regions for αSyn inclusions that are generally considered pathological by being proteinase K (PK)-resistant and immunopositive for pSer129-αSyn. When we examined the 9-month-old αSyn transgenic mice, we found that both experimental groups (i.e., those who were on DSS and those who stayed on normal water throughout their entire life and thus never experienced DSS colitis) exhibited extremely low levels of pathological αSyn aggregation in the brain (Fig. 5 and Suppl. Fig. 5). Our observation of the level of pathological αSyn aggregations in the brain of these 9-month-old hemizygous (Thy1)-h[A30P]αSyn transgenic mice (Fig. 5A) is indeed consistent with earlier descriptions of the model at the age of 11 months [57]. The 21-month-old hemizygous (Thy1)-h[A30P]αSyn transgenic mice that only received water during their lifetime showed, in contrast to the 9-month-old cohort, more discernible PK-resistant pSer129-αSyn immunoreactive features (Fig. 5B) and the abundance of these features was consistent with previous observations in this transgenic line at the age of 24 months [57]. In marked contrast, the 21-month-old hemizygous (Thy1)-h[A30P]αSyn transgenic mice that were exposed to DSS at three months of age presented with pSer129-positive αSyn pathology throughout various brain regions in a much more exacerbated fashion than mice that were aged up to 21 months without having experienced DSS colitis at young age (Fig. 5B-E). The degree and distribution of PK-resistant αSyn in the brain was similar to what was previously described for homozygous (Thy1)-h[A30P]αSyn transgenic mice at the age of 8 to 9 months [57]. The significant aggravation of αSyn pathology in the substantia nigra (p ≤ 0.01 in a negative-binomial mixed-effects model adjusting for multiple comparisons performed over all brain areas) was accompanied by a significant loss of tyrosine hydroxylase (TH) and Nissl-positive cells at 21 months of age (p ≤ 0.05, Student’s t-test; Fig. 6). Together, we found that experimental DSS colitis at a young age caused an age-dependent exacerbation of PK-resistant pSer129-αSyn pathology and a loss of nigral dopaminergic neurons in the brains of (Thy1)-h[A30P]αSyn transgenic mice.
Fig. 5 A 23-days chronic DSS colitis insult at young age causes an age-dependent accumulation of proteinase K-resistant pSer129-αSyn in various brain regions of (Thy1)-h[A30P]αSyn transgenic mice. A 23-days chronic increasing dose DSS paradigm was performed with 3-month-old (Thy1)-h[A30P]αSyn transgenic mice. After recovering and further aging, various brain regions were analyzed for proteinase K (PK)-resistant pSer129-αSyn immunoreactivity in 9-month (A) and 21-month-old (B) mice, respectively. The dark brown features in (A) (barely any visible in both the water and the DSS group) and (B) (strongly visible with typical neuritic and punctated inclusion-type morphology) indicate PK-resistant inclusions with pSer129-αSyn immunoreactivity. Densitometric quantification of pSer129-αSyn immunoreactivity in different brain regions of 21-month-old mice (C, D) (n=6 mice per group). In order to visualize better the differences between the water versus the DSS group at 21 months, brain regions with large increase were plotted on an y-axis up to 250 A.U./px2 and small increase on a y-axis up to 40 A.U./px2. The about <5 to 150 A.U./px2 average values in the DSS group versus the about 0 to 30 A.U./px2 average values in the water group in several brain regions confirm the visual impression in panel (B). One 21-month-old mouse in the DSS group was excluded from analysis due to presumed failed treatment; it is included in the graphs. Statistical analyses were performed using negative-binomial mixed-effects models adjusting for multiple comparisons. E Representative heatmap of the average distribution scores of pSer129-αSyn immunoreactivity for each treatment group in varying brain regions in all the 9-month (A and Supplemental Figure 5) and 21-month-old (B) mice was generated in a sagittal mouse brain (n=10 mice per group). Statistical analyses were performed using linear mixed-effects model adjusting for multiple comparisons. A.U./px2, = mean grey value x area stained/total area assessed. Scale bars: 500 μm. Discussion Currently, there is no therapy for PD available to slow or stop disease progression and an obstacle in the quest to develop one is that we do not understand how the disease develops [58]. Abnormal intraneuronal accumulation of αSyn (i.e., in Lewy bodies and neurites) is a key neuropathological hallmark and the distribution of Lewy pathology in postmortem brain is used for staging in PD [2,59]. Accumulation of αSyn has also been observed in the peripheral nervous system in PD, some individuals at risk of developing the disease, and normal individuals [60–62]. Similar to this finding in humans, αSyn-immunoreactive inclusions and signs of age-dependent αSyn-related pathological changes have also been detected in the ENS of wild type rats [63,64] and several transgenic mouse models prior to development of brain pathology [21,65]. Based on preclinical models employing injection of brain extracts or re-combinant αSyn fibrils to different brain regions and intestines [15–17,19,20,46,66,67] together with postmortem brain pathology [56,59,68], it has also been suggested that αSyn pathology propagates temporospatially from cell-to-cell in a prion-like manner [3,59,66,68,69]. However, the initial factors triggering αSyn aggregation in the tissue or organ of origin of the pathology are yet to be established [58] and the involvement of peripheral stimuli in the aggregation and pathogenic spread of αSyn is only beginning to unravel. In this study, we provide evidence that DSS colitis, i.e., an experimental IBD-like inflammation, triggers αSyn accumulation in the ENS of wild type mice and in a human αSyn transgenic mouse model of PD (Fig. 2). We found aggravation of enteric αSyn accumulation in αSyn transgenic mice lacking Cx3cr1 signaling and amelioration of inflammation and associated slight reduction of enteric αSyn load by systemic IL-10, demonstrating that genetic and pharmacologic modulation of inflammation can influence the degree of αSyn accumulation in the ENS (Fig. 3 and 4). Because IL-10 and the CX3CR1-CX3CL1 axis are able to mediate this effect, this suggests that monocytes/macrophages may modulate the process in this model. We further observed that the aggravated αSyn accumulation in the ENS persisted even after two months of recovery from DSS colitis and was aggravated in the absence of CX3CR1 signaling. This indicates that the accumulation is persistent and this further establishes that monocytes/macrophages play a critical role in this process (Suppl. Fig. 4). Remarkably, at 18 months but not 6 months post induction of DSS colitis (thus, at age 21 months but not 9 months, respectively), αSyn transgenic mice had developed massively elevated αSyn brain pathology (Fig. 5 and Suppl. Fig. 5). This elevated PK-resistant pSer129-αSyn pathology in the midbrain, including the substantia nigra, and other brain regions coincided with an average decrease of 30-50% of TH- and Nissl-positive cells in the nigra (Fig. 6). We chose to perform the long-term experiments in αSyn transgenic mice rather than wild type mice while being aware of the caveats of employing genetic overexpression models, which use a neuron selective Thy-1 promoter cassette such as these (Thy1)-h[A30P]αSyn transgenic mice. However, these particular αSyn transgenic mice had previously been shown to slowly develop αSyn pathology in the brain on a homozygous genotype [41,57] making them ideal when asking the question of whether transient colonic inflammation can aggravate brain pathology in a genetically predisposed animal such as the hemizygous transgenic mice used in this study. Others have recently demonstrated in a more aggressive αSyn transgenic mouse model that mild DSS colitis can accelerate αSyn accumulation in the ENS and brain [38]. In future long-term studies, we plan to address whether αSyn pathology develops also in the brains of wild type mice if they sustain transient experimental IBD at a young age. In our present study, experimental DSS colitis in αSyn transgenic mice recapitulated the accumulation of enteric αSyn which is proposed to occur in humans several years before PD diagnosis [39]. Additionally, the subsequent age-related development of αSyn pathology in the brain of αSyn transgenic mice together with the loss of nigral dopaminergic neurons mimicked a progression of the disease similar to what is considered to occur in PD.
Fig. 6 A 23-days chronic DSS colitis insult at young age results in loss of tyrosine hydroxylase- and Nissl-positive cells in the substantia nigra of (Thy1)-h[A30P]αSyn transgenic mice at 21 months of age. We established that a mechanism by which a specific type of peripheral inflammation promotes αSyn accumulation in the colon potentially involves monocytes and macrophages. Both peroral DSS and intraperitoneal LPS administration provoked strong local immune reactions resulting in leukocyte infiltration into the submucosa of the colon. The inflamed region of the colon contains the submucosal plexus and is anatomically separated from the myenteric plexus by a thick circular muscle (Fig. 1). This discrete localization of inflammation to the submucosa might explain why αSyn only accumulated in the nerves of the submucosal plexus and not in the myenteric plexus of our mice that received DSS in both a strong acute and the two chronic paradigms. The mechanism underlying how intraperitoneally administered LPS leads to submucosal leukocyte infiltration likely involves the monocyte attractant chemokine CCL2 (Fig. 3), but the specifics remain to be clarified [70]. Indeed, CCL2 was also upregulated in the colon of our DSS model. However, in contrast to intraperitoneal LPS, where infiltrating macrophages were present in discrete patches in the colonic wall, DSS-related macrophage infiltration was distributed both in small groups and larger randomly distributed patches of cells across the entire colonic submucosa. Also, perorally administered DSS destroys the mucosa of the colon, similar to some forms of ulcerative colitis, resulting in the transient disintegration of the intestinal epithelial barrier. In our (Thy1)-h[A30P]αSyn transgenic mice, the subsequent immune response to the infiltration of commensal bacteria evoked an elevated expression of cytokines such as IL-1β and IL-6, a phenomenon also observed in the colon of IBD patients [71,72]. This upregulation was absent in the LPS paradigm in which the intestinal mucosa remained intact. By acting on tight junctions, IL-1β and IL-6 can increase intestinal barrier permeability (gut leakiness), facilitating the recruitment of additional immune cells to the site of the inflammation, eventually culminating in widespread immune activation [73,74]. Consistent with the breach of barrier permeability in our mouse model, some PD patients exhibit increased colonic cytokines such as IL-1β, IL-6 and TNF, occurring together with increased intestinal permeability [23,75]. In this context, it is also notable that people with Crohn’s disease present with increased enteric αSyn expression [55] and even more striking that IBD patients on anti-TNF therapy have a reduced risk of developing PD compared to IBD patients not given this treatment [35]. Notably, mucosal macrophages with intra-lysosomal αSyn content were previously described in the intact human appendix [76]. These macrophages were in close proximity to the axonal varicosities of the vermiform appendix, which showed an enriched staining for αSyn in the mucosal plexus. Furthermore, we recently found that the vermiform appendix contains aggregated and truncated αSyn that has the propensity to seed aggregation of recombinant αSyn in vitro [62]. Our finding of LPS not being able to induce an αSyn phenotype in the colon is in contrast to the reports by Kelly and colleagues who injected wild type mice with LPS and observed an immediate and progressive increase in αSyn immunoreactivity in the myenteric ganglia of the large intestine but not in the small intestine [77]. The low dose (0.5 mg/kg used in our study versus 2.5 mg/kg used by Kelly and colleagues), coupled with the possible usage of different bacterial strains to generate LPS affecting potency and pyrogenicity of LPS (here strain O55:B5 was administered versus presumably strain O111:B4 used by Kelly and colleagues and others [78], and potential effects of different environments and microbiomes [79] may contribute to this discrepancy. It will be interesting to study the mentioned parameters and their role in inducing αSyn pathology further in rodent models and explore how this could be translated to the even more heterogeneous human setting. Similarly, others showed that employing a sub-chronic 0.5% DSS paradigm for 3 weeks in wild type mice did not trigger an αSyn pathology in the colon [80]. While we have not tested this particular sub-chronic DSS colitis paradigm and instead had administered 5% DSS acutely to the wild type mice (Fig. 2C), the data by Garrido-Gil and colleagues may corroborate our results which show in αSyn transgenic mice that a certain DSS dose and severity of inflammation is required in order to induce accumulation of αSyn in the submucosal plexus (Fig. 2A). What could be a functional role of the αSyn species found in abundance in the gut wall? Monomeric and oligomeric αSyn species reportedly act as chemoattractants for neutrophils and monocytes, enhancing the maturation of dendritic cells in the ENS [22,81]. With such a role in intestinal immunity, it is possible that the tissue destruction induced by DSS in the present study led to release of αSyn, which perhaps served as a chemoattractant for monocytes. The increased abundance of αSyn and altered intestinal permeability, along with the DSS-evoked inflammatory response may have provided an enabling milieu allowing further αSyn accumulation in the ENS of the colon [77]. Macrophages and other immune cells are also regulated by several genes including LRRK2, an established risk gene for PD and IBD. It will be interesting to explore how mutations in genes that control autophagy, including the LRRK2 gene, influence the handling of αSyn by macrophages that invade the inflamed colon in our DSS colitis paradigm. Despite the intriguing translational aspect of our finding in the DSS paradigm, others have very recently reported that DSS colitis in mice down-regulates the expression of enteric αSyn on protein levels in vivo [82,83]. This is in contrast to our immunofluorescence (i.e., increased accumulation of αSyn in submucosal plexus upon DSS colitis; Fig. 2, 3, and 4) and gene expression data (e.g., no change in endogenous and transgenic αSyn upon DSS colitis; Suppl. Fig. 3) in the same paradigm and may reflect the well-known lab-to-lab variability that can occur for the DSS models [84]. Perhaps the most striking finding in our study was that a single period of DSS-induced colitis at a young age led to an exacerbation of αSyn pathology in the brain of αSyn transgenic mice much later in life (Fig. 5). How does severe αSyn inclusion pathology develop in the brains of these mice? One hypothesis is that the brain αSyn pathology observed in this study could be due to direct effects of peripheral immune activation on the brain and that certain peripheral triggers can directly affect microglial activity. For instance, short-chain fatty acids derived from gut microbiota appear to influence function and maturation of microglia in the mouse brain [85] and inflammatory mediators released by gut microbiota into the bloodstream have been suggested to induce brain pathology and behavioral changes in an αSyn transgenic mouse model [86]. Moreover, rats and nematodes have been reported to develop αSyn inclusions after exposure to the bacterial amyloid protein curli, a protein which stimulates microgliosis, astrogliosis, and secretion of IL-6 and TNF [87]. Intriguingly, a recent study reported that peripherally applied inflammatory stimuli induce acute immune training (that exacerbates β-amyloid pathology) and immune tolerance in the brain that reprograms microglia, an effect which can persist for at least six months [88]. Whether this is a relevant mechanism in the DSS paradigm needs to be explored. Another hypothesis is that the observed brain αSyn pathology may have accumulated as a consequence of the transfer of pathogenic αSyn seeds from the gut via the vagal nerve. Several experimental studies have demonstrated that pathogenic αSyn seeds can be transferred from the peripheral to the central nervous system. Aggregated recombinant αSyn injected intraperitoneally, intramuscularly or into the gastric wall of certain mouse models of PD results in αSyn inclusions in the brain [16,89]. Data from animals injected with recombinant αSyn protein in the gut wall or viral vectors expressing αSyn into the vagal nerve suggest that pathogenic seeds can be transmitted via the vagal nerve [15,19,20,64,67,90–92]. A role for the vagal nerve in PD was also suggested by an epidemiological study indicating that vagotomy in a Danish population is associated with decreased PD risk [93], although this association has been challenged [94]. In the present study, αSyn pathology was much more prominent in the reticular nucleus (including the vagal nucleus) and midbrain areas compared to the rostral areas at 18 months post DSS colitis. Although we did not conduct the definitive experiment of cutting the vagal nerve, our data are consistent with the growing body of evidence that the vagal nerve is involved in the accumulation of αSyn aggregates in the brain. That said, the innervation of the colon occurs via both parasympathetic (e.g., vagal output neurons) and sympathetic (e.g., in the celiac ganglion of the upper abdomen) nerves. The possibility of propagation of αSyn pathology via the two routes is supported by the observation that injection of recombinant αSyn fibrils to the duodenum of certain αSyn transgenic rats leads to accumulation of pathological αSyn in organs innervated by the parasympathetic and the sympathetic nerves [67] and an age-dependent propagation of αSyn pathology from gut-to-brain in wild type rats [64]. Clinically, more relevant, αSyn accumulation is present in both parasympathetic and sympathetic vagal nerves in humans as well [95]. Thus, propagation may occur through both vagal and spinal routes. Aging is considered a major risk factor for neurodegenerative diseases, as failing cellular mechanisms are proposed to be unable to efficiently clear pathologically accumulating proteins and organelles [96]. Intriguingly, in the current study, while both cohorts were exposed to the same peripheral DSS colitis insult at 3 months of age, αSyn pathology only developed in the brain of aged (21-month-old) but not in the relatively younger (9-month-old) mice (Fig. 5). While the mechanisms involved in this phenomenon are still unclear, a recent study in mice reports an age-dependent association of development of αSyn pathology in the brain and development of motor deficits after inoculating the gut wall with recombinant αSyn fibrils [20]. Investigating this further, the study observed a reduction of the lysosomal enzyme glucocerebrosidase in the gut of aged but not young mice [20]. Viral vector-based overexpression of glucocerebrosidase partially rescued the ENS network connectivity with concomitant variable downregulation of pSer129-αSyn levels [20]. Although other age-related factors are likely involved, the finding of Challis and colleagues, together with the well-known genetic risk for glucocerebrosidase and several other lysosomal genes in PD [97,98], suggest a critical role of the lysosomal pathway in the long-term persistence of pathology in the ENS and the progression of αSyn pathology from the gut to the brain of aged mice. Upcoming studies in human tissue may shed more light on the mechanisms on how autophagy-lysosomal pathways are regulated and how they, together with aging, may contribute to the pathogenesis in PD. In summary, we report that αSyn accumulates in the colon of αSyn transgenic and wild type mice subjected to experimental DSS colitis and that this process can be modulated by genetically and pharmacologically modifying pathways related to monocyte/macrophage signaling. We further demonstrate that chronic but transient DSS colitis in young αSyn transgenic mice leads to a markedly exacerbated accumulation of αSyn aggregates in the brain of aged mice. In the same aged mice, the numbers of TH- and Nissl-positive neurons in the substantia nigra are reduced, suggestive of a neurodegenerative process. Together, our findings are in consonance with studies demonstrating a link between IBD and PD [33,35,99] and suggest a critical role for specific types of intestinal inflammation and αSyn accumulation in the initiation and progression of PD. Acknowledgments including sources of support We thank Drs. L. Ozmen and A. Bergadano for their tremendous support in maintaining the mouse colony and we are grateful to the animal caretakers, veterinarians and many unnamed staff at Roche for their valuable work with the mice in this study. In addition, at Roche we thank Dr. K.G. Lassen for critical input to the paper, Dr. C. Ullmer for co-mentoring S.G. and providing scientific input, Dr. L. Collin for helping with confocal imaging and we are grateful to Dr. T. Kremer, N. Haenggi, D. Mona, A. Girardeau, and J. Messer for providing support in tissue dissections and G. Walker and R. Lauria for technical support. Ms. E. Schulz from VAI assisted with immunostaining of the brain tissue. We thank the Contract Research Organization Frimorfo for carefully sectioning the brains for this study. We acknowledge Drs. L. Gaudimier (née Chicha) and F. Pan-Montojo for scientific discussions early in the project, Dr. W. Zago from Prothena for valuable scientific input throughout the project, and Drs. M. and P. Derkinderen for critical input on the link between IBD and the enteric αSyn accumulation in humans. P.B. acknowledges the Van Andel Institute and the many individuals and corporations that supported financially the neurodegenerative research at Van Andel Institute. Research at Van Andel Institute reported in this publication was also financially supported by Roche through a research collaboration with P.B. S.G and N.M. were supported by a grant from Roche under the Roche Postdoctoral Fellowship (RPF) Program. Conflict of interest At the time of the study S.G. and N.M. were Roche Postdoctoral Fellows employed by Roche and L.S., F.B., G.D.P., J.S.P., K.O.S., H.R., M.H., M.Se. M.St., P.M., A.W., T.E., A.H. and M.B. are or were fulltime employees or trainees at Roche and they may additionally hold Roche stock/stock options. S.G. and L.S. are currently employees of Neurimmune AG, Schlieren, Switzerland. P.B. has received commercial support as a consultant from Axial Biotherapeutics, Calico Life Sciences, CuraSen, Fujifilm-Cellular Dynamics International, H. Lundbeck A/S, and Idorsia Pharmaceuticals Ltd. He has received commercial support for grants/research from H. Lundbeck A/S and Roche. He has ownership interests in Acousort AB. The other authors declare that they have no competing interest with regard to this research. Author contributions S.G., N.M. and L.S. planned and performed the in vivo experiments, colon immunostaining, analysis, and quantification; S.G. and N.M. drafted a first version of the manuscript; E.Q. performed, imaged, quantitated pSer129, TH and Nissl staining in the brain sections, and drafted a more advanced version of the manuscript with J.A.S., who also provided helpful discussion. Both J.A.S. and E.Q. were critical in revising the manuscript. F.B. and K.O.S. supported the image acquisition and image analysis for the colon samples; M.St. performed imaging and data analysis of experiments with wild type mice; G.D.P. and J.S.P. performed statistical analysis of the DSS experiments; H.R. and M.H. performed mRNA analyses; M.Se. trained S.G. and L.S. on mouse necropsy and supported their work; P.M. performed expert pathology staging on leukocyte infiltration; T.E. and A.W. provided mIgG-mIL-10 fusion proteins and measured serum exposure; Z.M. performed statistical analysis for the pSer129 αSyn immunohistochemistry data. A.S. contributed with scientific and veterinary expert input for implementation and analysis of the DSS colitis model at Roche. M.L.E.G. provided helpful discussion and project planning. A.H. co-mentored S.G. and N.M., performed expert pathology staging on leukocyte infiltration and contributed to experimental planning. C.M. trained S.G. on the colitis model and provided expert input on the experimental IBD model. M.B. and P.B. co-mentored Roche Postdoctoral Fellows S.G. and N.M., conceived and oversaw the study, and performed experimental planning; M.B., P.B. J.A.S. and E.Q. wrote the final version of the manuscript. All authors read and approved the final manuscript. References 1. Tysnes O-B, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm (Vienna). 2017;124:901–5. 2. Spillantini MG, Goedert M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2017;373:137–48. 3. Del Tredici K, Braak H. Lewy pathology and neurodegeneration in premotor Parkinson’s disease. Mov Disord. 2012;27:597–607. 4. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. 5. Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14:57–64. 6. Gaenslen A, Swid I, Liepelt-Scarfone I, Godau J, Berg D. The patients’ perception of prodromal symptoms before the initial diagnosis of Parkinson’s disease. Mov Disord. 2011;26:653–8. 7. Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov Disord. 2015;30:229–37. 8. Berg D, Godau J, Seppi K, Behnke S, Liepelt-Scarfone I, Lerche S, et al. The PRIPS study: screening battery for subjects at risk for Parkinson’s disease. Eur J Neurol. 2013;20:102–8. 9. Postuma RB, Gagnon J-F, Bertrand J-A, Génier Marchand D, Montplaisir JY. Parkinson risk in idiopathic REM sleep behavior disorder: preparing for neuroprotective trials. Neurology. 2015;84:1104–13. 10. Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–62. 11. Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, et al. Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology. 2009;73:1752–8. 12. Mahlknecht P, Seppi K, Poewe W. The Concept of Prodromal Parkinson’s Disease. J Parkinsons Dis. 5:681–97. 13. Braak H, de Vos RAI, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. 14. Phillips RJ, Walter GC, Wilder SL, Baronowsky EA, Powley TL. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson’s disease? Neuroscience. 2008;153:733–50. 15. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–20. 16. Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G. Neuroinvasion of α-Synuclein Prionoids after Intraperitoneal and Intraglossal Inoculation. J Virol. 2016;90:9182–93. 17. Sargent D, Verchère J, Lazizzera C, Gaillard D, Lakhdar L, Streichenberger N, et al. “Prion-like” propagation of the synucleinopathy of M83 transgenic mice depends on the mouse genotype and type of inoculum. J Neurochem. 2017;143:126–35. 18. Manfredsson FP, Luk KC, Benskey MJ, Gezer A, Garcia J, Kuhn NC, et al. Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis. 2018;112:106–18. 19. Arotcarena M-L, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. Oxford Academic; 2020;143:1462–75. 20. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nature Neuroscience. Nature Publishing Group; 2020;23:327–36. 21. Bencsik A, Muselli L, Leboidre M, Lakhdar L, Baron T. Early and Persistent Expression of Phosphorylated α-Synuclein in the Enteric Nervous System of A53T Mutant Human α-Synuclein Transgenic Mice. Journal of Neuropathology & Experimental Neurology. 2014;73:1144–51. 22. Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S, et al. A Role for Neuronal Alpha-Synuclein in Gastrointestinal Immunity. JIN. 2017;9:456–63. 23. Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–8. 24. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–50. 25. Brás J, Guerreiro R, Hardy J. SnapShot: Genetics of Parkinson’s disease. Cell. 2015;160:570-570.e1. 26. Reynolds RH, Botía J, Nalls MA, Hardy J, Taliun SAG, Ryten M. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. npj Parkinson’s Disease. 2019;5:6. 27. Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017;74:780–92. 28. Foo JN, Chung SJ, Tan LC, Liany H, Ryu H-S, Hong M, et al. Linking a genome-wide association study signal to a LRRK2 coding variant in Parkinson’s disease. Mov Disord. 2016;31:484–7. 29. Umeno J, Asano K, Matsushita T, Matsumoto T, Kiyohara Y, Iida M, et al. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2407–15. 30. Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu N-Y, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Science Translational Medicine. 2018;10:eaai7795. 31. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–85. 32. Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna). 2011;118:795–808. 33. Lin J-C, Lin C-S, Hsu C-W, Lin C-L, Kao C-H. Association Between Parkinson’s Disease and Inflammatory Bowel Disease: a Nationwide Taiwanese Retrospective Cohort Study. Inflamm Bowel Dis. 2016;22:1049–55. 34. Nerius M, Doblhammer G, Tamgüney G. GI infections are associated with an increased risk of Parkinson’s disease. Gut. 2019;gutjnl-2019-318822. 35. Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, et al. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol. 2018;75:939–46. 36. Rolli-Derkinderen M, Leclair-Visonneau L, Bourreille A, Coron E, Neunlist M, Derkinderen P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J Neurol. 2019;1–7. 37. Wan Q-Y, Zhao R, Wu X-T. Older patients with IBD might have higher risk of Parkinson’s disease. Gut. 2018;gutjnl-2018-317103. 38. Kishimoto Y, Zhu W, Hosoda W, Sen JM, Mattson MP. Chronic Mild Gut Inflammation Accelerates Brain Neuropathology and Motor Dysfunction in α-Synuclein Mutant Mice. Neuromolecular Med. 2019;21:239-249. 39. Houser MC, Tansey MG. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? npj Parkinson’s Disease. 2017;3:3. 40. Joers V, Masilamoni G, Kempf D, Weiss AR, Rotterman TM, Murray B, et al. Microglia, inflammation and gut microbiota responses in a progressive monkey model of Parkinson’s disease: A case series. Neurobiology of Disease. 2020;144:105027. 41. Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–73. 42. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–14. 43. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–53. 44. Schenk M, Bouchon A, Seibold F, Mueller C. TREM-1--expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest. 2007;117:3097–106. 45. Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–3. 46. Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, et al. Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 2017;1–19. 47. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. 48. Smyth GK. limma: Linear Models for Microarray Data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor [Internet]. New York, NY: Springer New York; 2005 [cited 2019 May 1]. p. 397–420. Available from: https://doi.org/10.1007/0-387-29362-0_23 49. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104:Unit 15.25. 50. Weber B, Saurer L, Schenk M, Dickgreber N, Mueller C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur J Immunol. 2011;41:773–9. 51. Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–95. 52. Kostadinova FI, Baba T, Ishida Y, Kondo T, Popivanova BK, Mukaida N. Crucial involvement of the CX3CR1-CX3CL1 axis in dextran sulfate sodium-mediated acute colitis in mice. J Leukoc Biol. 2010;88:133–43. 53. Kang S, Okuno T, Takegahara N, Takamatsu H, Nojima S, Kimura T, et al. Intestinal epithelial cell-derived semaphorin 7A negatively regulates development of colitis via αvβ1 integrin. J Immunol. 2012;188:1108–16. 54. Li B, Alli R, Vogel P, Geiger TL. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol. 2014;7:869–78. 55. Prigent A, Lionnet A, Durieu E, Chapelet G, Bourreille A, Neunlist M, et al. Enteric alpha-synuclein expression is increased in Crohn’s disease. Acta Neuropathol. 2019;137:359–61. 56. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. 57. Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–39. 58. Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2018;S0166-2236:30253–4. 59. Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol. 2002;249 Suppl 3:III/1-5. 60. Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord. 2012;27:716–9. 61. Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N’Guyen J-M, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE. 2010;5:e12728. 62. Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. 2018;10:eaar5280. 63. Phillips RJ, Walter GC, Ringer BE, Higgs KM, Powley TL. Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp Neurol. 2009;220:109–19. 64. Van Den Berge N, Ferreira N, Mikkelsen TW, Alstrup AKO, Tamgüney G, Karlsson P, et al. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain. 2021;awab061 65. Kuo Y-M, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated α-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–50. 66. Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. 67. Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138:535-550. 68. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. 69. Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, et al. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016;213:1759–78. 70. Puntambekar SS, Davis DS, Hawel L, Crane J, Byus CV, Carson MJ. LPS-induced CCL2 expression and macrophage influx into the murine central nervous system is polyamine-dependent. Brain Behav Immun. 2011;25:629–39. 71. Andus T, Daig R, Vogl D, Aschenbrenner E, Lock G, Hollerbach S, et al. Imbalance of the interleukin 1 system in colonic mucosa—association with intestinal inflammation and interleukin 1 receptor agonist genotype 2. Gut. 1997;41:651–7. 72. Sanchez-Muñoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–8. 73. Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178:4641–9. 74. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–71. 75. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLOS ONE. 2011;6:e28032. 76. Gray MT, Munoz DG, Gray DA, Schlossmacher MG, Woulfe JM. Alpha-synuclein in the appendiceal mucosa of neurologically intact subjects. Mov Disord. 2014;29:991–8. 77. Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RAE, et al. Progression of Intestinal Permeability Changes and Alpha-Synuclein Expression in a Mouse Model of Parkinson’s Disease. Mov Disord. 2014;29:999–1009. 78. Akarsu ES, Mamuk S. Escherichia coli lipopolysaccharides produce serotype-specific hypothermic response in biotelemetered rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1846-1850. 79. Dodiya HB, Forsyth CB, Voigt RM, Engen PA, Patel J, Shaikh M, et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol Dis. 2020;135:104352. 80. Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Bidirectional Neural Interaction Between Central Dopaminergic and Gut Lesions in Parkinson’s Disease Models. Mol Neurobiol. 2018;55:7297-7316 81. Labrie V, Brundin P. Alpha-Synuclein to the Rescue: Immune Cell Recruitment by Alpha-Synuclein during Gastrointestinal Infection. JIN. 2017;9:437–40. 82. Prigent A, Gonzales J, Durand T, Le Berre-Scoul C, Rolli-Derkinderen M, Neunlist M, et al. Acute inflammation down-regulates alpha-synuclein expression in enteric neurons. J Neurochem. 2019;148:746–60. 83. Resnikoff H, Metzger JM, Lopez M, Bondarenko V, Mejia A, Simmons HA, et al. Colonic inflammation affects myenteric alpha-synuclein in nonhuman primates. J Inflamm Res. 2019;12:113–26. 84. Whittem CG, Williams AD, Williams CS. Murine Colitis Modeling using Dextran Sulfate Sodium (DSS). J Vis Exp [Internet]. 2010 [cited 2019 Apr 27]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2841571/ 85. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–77. 86. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell. 2016;167:1469-1480.e12. 87. Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Scientific Reports. 2016;6:34477. 88. Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–8. 89. Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A. 2014;111:10732–7. 90. Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron [Internet]. 2019 [cited 2019 Jul 25];0. Available from: https://www.cell.com/neuron/abstract/S0896-6273(19)30488-X 91. Lohmann S, Bernis ME, Tachu BJ, Ziemski A, Grigoletto J, Tamgüney G. Oral and intravenous transmission of α-synuclein fibrils to mice. Acta Neuropathol. 2019;138(4):515-533. 92. Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Molecular Neurodegeneration. 2018;13:21. 93. Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78:522–9. 94. Tysnes O-B, Kenborg L, Herlofson K, Steding-Jessen M, Horn A, Olsen JH, et al. Does vagotomy reduce the risk of Parkinson’s disease? Ann Neurol. 2015;78:1011–2. 95. Braak H, Sastre M, Bohl JRE, de Vos RAI, Del Tredici K. Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007;113:421–9. 96. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Research Reviews. 2014;14:19–30. 97. Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132:1783–94. 98. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–93. 99. Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977-2014. Gut. 2018;68:18–24.
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |