|
|
|
Free Neuropathology 1:31 (2020) |
|
Review |
|
Aβ plaques |
|
Lary C. Walker |
|
Department of Neurology and Yerkes National Primate Research Center, Emory University |
|
Corresponding author: |
|
Submitted: 27 September 2020 Accepted: 23 October 2020 Copyedited by: Bert M. Verheijen Published: 30 October 2020 |
|
Keywords: Alzheimer’s disease, Amyloid, Neuritic plaques, Neurofibrillary tangles, Senile plaques |
|
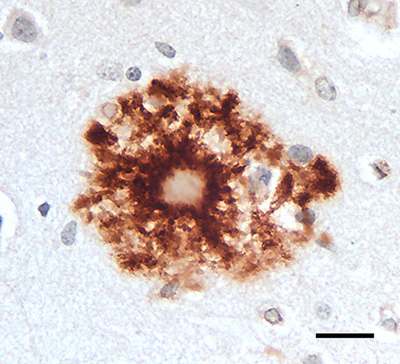
Abstract Aβ plaques are one of the two lesions in the brain that define the neuropathological diagnosis of Alzheimer’s disease. Plaques are highly diverse structures; many of them include massed, fibrillar polymers of the Aβ protein referred to as Aβ-amyloid, but some lack the defining features of amyloid. Cellular elements in ‘classical’ plaques include abnormal neuronal processes and reactive glial cells, but these are not present in all plaques. Plaques have been given various names since their discovery in 1892, including senile plaques, amyloid plaques, and neuritic plaques. However, with the identification in the 1980s of Aβ as the obligatory and universal component of plaques, the term ‘Aβ plaques’ has become a unifying term for these heterogeneous formations. Tauopathy, the second essential lesion of the Alzheimer’s disease diagnostic dyad, is downstream of Aβ-proteopathy, but it is critically important for the manifestation of dementia. The etiologic link between Aβ-proteopathy and tauopathy in Alzheimer’s disease remains largely undefined. Aβ plaques develop and propagate via the misfolding, self-assembly and spread of Aβ by the prion-like mechanism of seeded protein aggregation. Partially overlapping sets of risk factors and sequelae, including inflammation, genetic variations, and various environmental triggers have been linked to plaque development and idiopathic Alzheimer’s disease, but no single factor has emerged as a requisite cause. The value of Aβ plaques per se as therapeutic targets is uncertain; although some plaques are sites of focal gliosis and inflammation, the complexity of inflammatory biology presents challenges to glia-directed intervention. Small, soluble, oligomeric assemblies of Aβ are enriched in the vicinity of plaques, and these probably contribute to the toxic impact of Aβ aggregation on the brain. Measures designed to reduce the production or seeded self-assembly of Aβ can impede the formation of Aβ plaques and oligomers, along with their accompanying abnormalities; given the apparent long timecourse of the emergence, maturation and proliferation of Aβ plaques in humans, such therapies are likely to be most effective when begun early in the pathogenic process, before significant damage has been done to the brain. Since their discovery in the late 19th century, Aβ plaques have, time and again, illuminated fundamental mechanisms driving neurodegeneration, and they should remain at the forefront of efforts to understand, and therefore treat, Alzheimer’s disease. 1. Aβ, amyloid, and Alzheimer’s disease The most striking and yet still enigmatic pathologic features of Alzheimer’s disease (AD) are lesions known for over a century as senile plaques - microscopic anomalies in the parenchyma of the brain consisting of an abnormal accumulation of protein decorated by various molecules, and often including dystrophic neuronal processes and reactive glial cells (Figure 1). Although plaques are a frequent feature of the senescent brain and, when particularly numerous, an obligatory diagnostic marker of AD [1], the identity of the principal protein in the plaque core remained unknown until the 1980s. Then, Glenner and Wong established a partial amino acid sequence of the protein in cerebral amyloid angiopathy (CAA) from patients with AD and Down syndrome [2, 3], and Masters and Beyreuther [4, 5] determined that the same protein is a key component of plaques. Initially referred to as the β protein, A4, or β/A4, the protein now is commonly designated Aβ [6]. Collectively, these lesions are increasingly referred to as Aβ plaques (see Section 3).
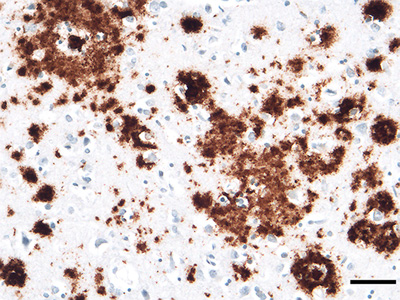
Figure 1. 'Classical' Aβ (senile) plaques in the cortex of persons who had died with Alzheimer's disease (AD). Left, a plaque stained with the Naoumenko-Feigin silver method and periodic acid-Schiff (PAS) counterstain; an amyloid core (dark pink) is surrounded by profuse abnormal neurites (black). Right, a plaque immunostained with antibody 4G8 to the Aβ protein (brown) along with a Nissl counterstain (blue); glial nuclei are visible in the region between the plaque core and outer corona, and within and surrounding the corona. Bar = 20μm for both panels. 1.1 Aβ Aβ is a cleavage product of the Aβ-precursor protein (APP), a 695–770 amino acid, single membrane-spanning protein that is strongly expressed in the nervous system [7, 8]. Aβ is generated mainly in endosomes, and its release into the extracellular space is influenced by synaptic activity [9]. To produce Aβ, APP is sequentially cleaved by the enzymes β-secretase [or β-amyloid cleaving enzyme (BACE)] and γ-secretase [8], resulting in Aβ proteins that are most often 40 or 42 amino acids in length (‘Aβ40’ and ‘Aβ42’), although many C-terminally and N-terminally variant and/or chemically modified Aβ fragments also occur [7, 10-16]. Different lengths of Aβ can derive from their differential excision from APP by secretases or from post-translational trimming of Aβ by exopeptidases [10]. Potential post-translational chemical modifications of Aβ include pyroglutamylation, racemization, isomerization, oxidation, phosphorylation, N-homocysteinylation, nitration, and glycosylation [11, 17-19] (see also Section 7, below). How post-translational modifications influence the process of protein aggregation in general remains poorly understood [20, 21]. Aβ40 is the isoform of Aβ that is most abundantly generated by neurons, but two C-terminal hydrophobic residues in Aβ42 augment its tendency to self-assemble into amyloid [7, 22]. As a result, more plaques are immunoreactive for Aβ42 than for Aβ40 (Figure 2), although the relative amounts of plaques stained for Aβ40 and Aβ42 vary.
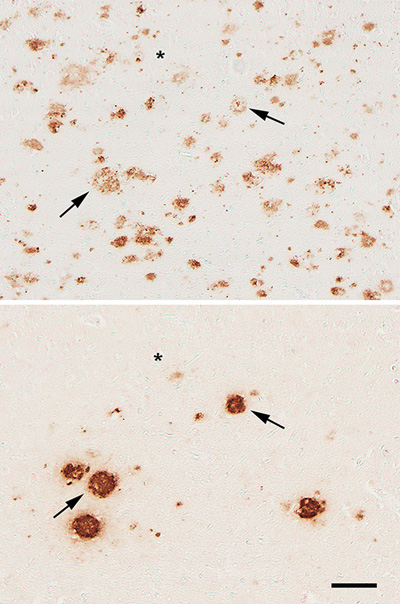
Figure 2. Adjacent cortical tissue sections from an AD patient, immunostained with antibodies R398 to Aβ42 (top) and R361 to Aβ40 (bottom). Two of the plaques that are present in both sections are denoted by arrows. Asterisks mark a blood vessel for reference. Bar = 100μm. Unlike plaques, cerebral Aβ-amyloid angiopathy (Aβ-CAA) in large vessels is more consistently positive for Aβ40, though Aβ42 also is generally present [23]. The staining patterns of the two isoforms differ in capillary Aβ-CAA compared to large-vessel Aβ-CAA [24, 25], and in the vessel wall compared to the diffuse Aβ that sometimes extends from the wall into the surrounding parenchyma (dyshoric amyloid angiopathy) [25, 26]. The mechanisms governing the ontogeny of plaques and Aβ-CAA also probably differ to some extent (see Section 5.3). In addition to plaques and amyloid angiopathy, Aβ multimerizes into a range of oligomeric species [27, 28] that can interact with cells and impair brain function [27, 29-35]. Oligomers appear to be an important intermediate step in the assembly of polymeric amyloid of all types [20]. Comparison of subjects expressing AD-type dementia to nondemented subjects with high Aβ plaque pathology, the amount of oligomeric Aβ correlates more strongly with cognitive decline than does the number of plaques per se [36]. Experimental studies indicate that Aβ plaques include abundant oligomers [36, 37], and that some plaques shed toxic oligomers into the surrounding parenchyma [37-39]. Aβ42-oligomers have been shown to arise from secondary nucleation on Aβ-amyloid fibrils during protein aggregation, directly linking them to the process of amyloidogenesis [34]. At least some Aβ-oligomers are particularly potent seeds for the formation of Aβ plaques [40, 41], although whether there are seed-active oligomers that differ from toxic oligomers, as has been found for prions [42], is unknown. The relationship between Aβ-oligomers and the diverse plaque types [31, 33, 38] in the human brain - e.g., dense-core vs diffuse - also is an issue that remains incompletely defined. Indeed, owing to their dynamicity and heterogeneity, the analysis of oligomers as they occur in living systems is technically challenging [20] (see also Benilova et al. [43] for a critique of oligomers as toxic agents). Regardless of the relative contribution of Aβ-oligomers and amyloid fibrils to disease, both of these multimeric states denote the presence of an abnormal condition in the brain, i.e., the misfolding and accumulation of the Aβ protein. Aβ has assumed a prominent position in Alzheimer research because all identified risk factors for AD increase its quantity and/or tendency to aggregate [33, 44, 45]. Most notably, mutations in APP and the presenilins (components of the γ-secretase complex) [22] are the only known autosomal dominant causes of AD, and a superfluous APP gene due to trisomic chromosome 21 in Down Syndrome frequently leads to early-onset AD [35, 46, 47]. Furthermore, a rare mutation that substitutes a threonine for alanine (A673T) at position 2 of Aβ lowers both the production of Aβ [48] and its propensity to aggregate [49]; this mutation is associated with a reduced risk of manifesting AD [48] and possibly parenchymal plaques as well [50]. Contrariwise, when a valine replaces alanine at position 2 (A673V), Aβ generation is increased, and the protein is more prone to aggregate, resulting in an autosomal recessive form of AD [51]. Thus, there is little doubt that Aβ is intimately involved in the pathogenesis of AD, although many questions remain about how plaques per se participate in the neurodegenerative process. 1.2 Amyloid A persistent source of misunderstanding regarding the role of Aβ in AD is the common use of the generic term ‘amyloid’ to refer to the protein Aβ. In pathology, amyloid refers to ‘mainly extracellular tissue deposits of protein fibrils, recognized by certain properties, such as green-yellow birefringence after staining with Congo red’ [6] (for historical considerations of amyloid, see [20, 52-55], and for more on the definition of amyloid see [20, 56, 57]. Amyloid can arise from over 30 different proteins in various parts of the body in different human diseases [6, 58]. Hence, ‘Aβ’ the molecule and ‘amyloid’ the fibrillar mass are not synonymous. Aβ refers exclusively to the protein that, when aggregated into distinctive fibrils, constitutes the specific type of amyloid that most commonly accumulates in the aging brain. The formation of amyloid involves the misfolding and self-assembly of a particular protein into filamentous structures with a distinctive cross-β architecture that is stabilized by a ‘steric zipper’ molecular motif [20, 59]. The misfolded protein has two notable characteristics that contribute to its amyloidogenicity: 1) it compels unfolded molecules of the same protein to similarly misfold by means of permissive templating [60]; and 2) the β-sheets in separate molecules hydrogen-bond to one another to form stable, filiform polymers with the β-sheets oriented perpendicular to the long axis of the polymer [59]. In this way, the misshapen proteins both corrupt and capture like proteins, which stack into protofilaments that wind together to build long, non-branching fibrils that typically range from ~6 to 13 nm in diameter [54]. These fibrils are characteristic of amyloid in general [6]. Despite their shared cross-β backbone and similar appearance by conventional transmission electron microscopy, however, amyloid fibrils are polymorphic at the molecular level [20, 61-68]. Although amyloid was long defined as an exclusively extracellular substance [69-71], it is now recognized to occur intracellularly as certain types of inclusion [6, 20]. The tau protein that polymerizes into neurofibrillary tangles - the second mandatory pathologic hallmark of AD (Figure 3) - has attributes of amyloid [72]. Thus, the two lesions that characterize AD pathologically - plaques and tangles - arise from two different proteins - Aβ and tau - both of which can misfold and self-assemble into amyloid.
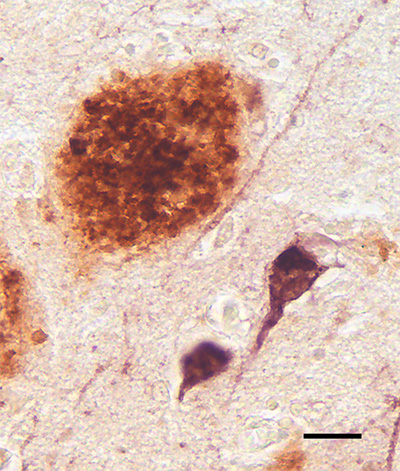
Figure 3. An Aβ plaque (brown) alongside intracellular tau tangles (purple) in the cortex of an AD patient. Combined polyclonal antibodies R398+R361 to Aβ40+42 plus monoclonal antibody CP13 to hyperphosphorylated tau. Bar = 20μm. Despite genetic, biomarker and pathologic findings implicating aberrant Aβ in the initiation of AD [9, 33, 44], tauopathy is more strongly correlated with cognitive decline than are plaques [73-78]. In the forebrain, tangles first appear in the medial temporal lobe [79, 80], but the dementia of AD is fully apparent only when tauopathy becomes severe in much of the neocortex [1, 81], a process that is facilitated by the presence of Aβ pathology [9] (see also [82]). The precise nature of the mechanistic link between Aβ-proteopathy and tauopathy in AD, however, remains a critical unsolved problem [45, 83, 84]. 2. The discovery and early exploration of plaques The late 19th and early 20th centuries saw a profusion of new staining methods that selectively revealed various elements in cells and tissues [85]. Accordingly, the original depictions of plaques reflected what was disclosed by histologic stains and viewed with the light microscope. In 1892, Paul Blocq and Georges Marinesco at the Salpêtrière Hospital in Paris reported microscopic ‘amas ronds’ (‘round clusters’, or ‘round heaps’) or ‘nodules de sclérose névroglique’ (‘nodules of neuroglial sclerosis’) in the brains of older epileptic patients [86].1 This report is generally considered to be the first unambiguous identification of plaques in the senescent brain [81, 87]. In 1898, Emil Redlich published evidence linking plaques to dementia [88]; in the brains of three elderly subjects, two of whom had died with clinically confirmed dementia, Redlich described the structures as consisting of a core of uncertain substance along with surrounding astrocytes (‘Spinnenzellen’) and their processes. Because they resembled millet seeds under the microscope, he referred to these collective lesions as ‘miliary sclerosis’ (‘miliare Sklerose’). Notably, Redlich also dubbed them ‘plaques’, a term that was expanded to ‘senile plaques’ by Simchowicz in 1911 [89]. Furthermore, Redlich noted that some smaller lesions consisted of fine fibers with a cotton-like appearance [88], anticipating the use of the term ‘cotton-wool plaques’ to depict certain types of deposit today [90-94]. Although Alois Alzheimer is often credited with instigating the burst of scientific analyses of plaques with his 1906 conference presentation in Tübingen (published in 1907) [95], his report was brief, and plaques (‘miliary foci’) were only superficially mentioned.2 He did not issue his first detailed histopathologic examination of plaques until 1911 [96]. In fact, along with Redlich [88], a good case can be made that Oskar Fischer deserves the credit for initiating the modern histopathologic analysis of dementia with a comprehensive series of reports published in 1907, 1910, and 1912 [87]. Several other researchers contributed to the literature on plaques during this period, including, among others, Miyake [97], Lafora [98], Bonfiglio [99], Hübner [100], Perusini [101], Fuller [102], Bielschowsky [103], Barrett [104], Simchowicz [105] and Marinesco and Minea [106] (see also Christen [107] for a brief historical perspective on this period of research into what we now call AD). Both Alzheimer and Fischer excelled in their analysis of plaques by implementing a silver-based staining method introduced by Max Bielschowsky [87] (see Braak and Braak [108] for a nice summary of the early development of silver stains).3 Alzheimer did, however, correctly anticipate the evolution of neurology in his 1907 publication, in which he contended that the time had come to define neurologic diseases based on both their clinical and histologic characteristics [95].4 This view has a contemporary parallel in the call by an international group of experts for a biological, rather than syndromic, definition of AD [109]. Furthermore, Alzheimer noted in 1911 the prevailing technical inability to identify the substance in the plaque core: ‘... we have to consider the core of the plaque as an unorganized mass which emerges differently with different staining methods ... As Perusini and Fischer have already explained, we are not at present able to identify this mass with any of the substances known in pathological anatomy’ (translation from [110]). In addition, Alzheimer highlighted the prominence of glial cells in the composition of plaques [96], a subject that has gained momentum in the 21st century, owing in part to the identification of compelling, glia-related genetic risk factors for AD [111-115] (see Section 6.2). From the early 20th century on, researchers widely agreed that the main structural elements comprising plaques are abnormal neuronal processes, altered glial cells, and a central, disordered mass of unidentified material. In a 1929 review, Macdonald Critchley [116] wrote that the ‘modern conception of the plaque is that of a reactionary change directed against a specific metabolic process of a toxic nature’ (a description that, if we consider the material in the core to be the key toxic substance, resonates with leading 21st century concepts). Many pioneering scientists attempted to explain the origin and nature of plaques based on their interpretations of static images in selectively stained tissue sections. Not surprisingly, disagreement was common (see, e.g., [99], [101], [102], [117]). Ferraro (1931) summarized this lack of consensus: ‘...one group of investigators favors the theory that [the plaque] originates from nerve cells, another that it originates from neuroglial elements, another from axis cylinders, and still another, from the intercellular ground substance’ [118]. Soniat remarked in 1941 that ‘No less than twenty different concepts concerning their origin have appeared in the literature’ [119]. As late as 1960, Liss wrote, citing three influential textbooks on pathology, that the ‘morphogenesis of senile plaques remains still an unsettled and controversial matter’ [120]. A crucial question, and a source of much of the discord among researchers, was the nature of the plaque core - what does it consist of, how does it arise, what impact does it have, and what governs the proliferation of plaques in the brain? The answers to these questions would not begin to emerge for another half century. In fact, no compelling conceptual insights immediately followed the initial flurry of histopathological investigations of plaques, which, ultimately, were hampered by limitations in the available methods [119, 121]. Beginning in the 1960s, theoretical and analytical advances accelerated; electron-microscopic studies showed that the mature plaque core consists of amyloid fibrils structurally similar to those in corporeal amyloidoses [122, 123], and quantitative analyses confirmed that plaque load in the brain is linked to dementia [124]. Most important, however, was the molecular decipherment of Aβ as the primary protein in cerebral amyloid by Glenner and Wong [2, 3] and Masters and colleagues [4]. The genetic insights and technical tools resulting from this discovery ultimately established Aβ as a critical player in the pathogenesis of AD, and the plaques that occur in normal aging and AD could, for the first time, be unified by a single, omnipresent component - aberrant Aβ. 1 ‘Il existe de plus, disséminés dans les diverses couches de l’écorce, de petits amas ronds d’un diamètre de 60 µ environ, se distinguant du reste du tissu par une coloration beaucoup plus intense, à contours réguliers. Ils apparaissent ainsi, parsemant discrètement le fond des préparations, d’une structure vaguement pointillée, ce pourquoi il est permis de considérer quelques-uns d’entre eux, au moins, comme de véritables nodules de sclérose névroglique (?).’ (Question mark is in the original) 2 ‘Über die ganze Rinde zerstreut, besonders zahlreich in den oberen Schichten, findet man miliare Herdchen, welche durch Einlagerung eines eigenartigen Stoffes in die Hirnrinde bedingt sind. Er lässt sich schon ohne Färbung erkennen, ist aber Färbungen gegenüber sehr refractär.’ 3 Many different silver stains have been developed to detect AD pathology. Each method selectively reveals certain elements in the plaques, and they are sometimes considered to be less sensitive than is immunostaining with antibodies to Aβ. Some silver stains, however, are exquisitely sensitive even to small, diffuse Aβ deposits, which have been recognized in AD tissue since the early 20th century (see, e.g., Marinesco and Minea [1912] [reference 106] and Cowe, A. [1915] [reference 508]). Note also Figure 23. 4 ‘Es gibt ganz zweifellos viel mehr psychische Krankheiten, als sie unsere Lehrbücher aufführen. In manchen solchen Fällen wird dann eine spätere histologische Untersuchung die Besonderheit des Falles feststellen lassen. Dann werden wir aber auch allmählich dazu kommen, von den großen Krankheitsgruppen unserer Lehrbücher einzelne Krankeiten (sic) klinisch abzuscheiden und jene selbst klinisch schärfer zu umgrenzen.’ 3. Plaque nomenclature: The case for ‘Aβ plaques’ The term ‘plaque’ (which historically has referred to a flat object such as a disk or tablet) was adopted by the medical community in the mid-to-late late 1800s to designate patch-like abnormalities such as atherosclerotic plaque or dental plaque.5 Redlich [88] used the term to describe carmine-stained densities (‘intensiv gefärbten Plaques’), and Simchowicz [105] added the modifier ‘senile’ to denote their frequency in senescent brains, particularly in patients with senile dementia [89]. Most plaques in the brain (unlike dental or atherosclerotic plaque) are not planar (one exception being the band-like subpial deposits [see Figure 9]). Of course, spheroidal plaques appear discoid in histologic sections, and their apparent size and composition are influenced by the plane through which they are cut (Figure 4).
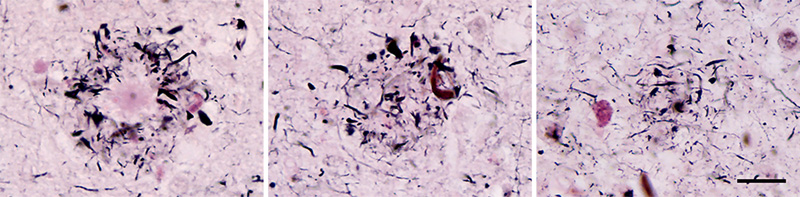
Figure 4. A neuritic Aβ plaque in consecutive sections of the cortex from an AD patient; The core is evident in the left-hand image, whereas sections through the periphery (middle and right) reveal only neurites (black). Serial sections may be required to unequivocally identify plaque types (a technical caveat noted by, among others, Alzheimer [1911] [reference 96]). Naoumenko-Feigin (silver) and periodic-Schiff stains. Bar = 20 μm for all images. Some of the designations for plaques derive from their staining characteristics. Following Divry’s discovery that certain plaques show amyloid-type birefringence after staining with the dye Congo red [125], the terms ‘congophilic plaques’ or ‘amyloid plaques’ became common. The term ‘argyrophilic plaques’ also has been employed, owing to their detectability by various silver-based staining methods [81]. Other labels such as ‘miliary plaques’, ‘Drusen’,6 and ‘Redlich-Fischer plaques’ can be found in the earlier literature [116]. In 1972, Wisniewski and Terry introduced the term ‘neuritic plaques’ in recognition of the profusion of abnormal neuronal processes that invest many plaques. With the identification of the Aβ protein in plaques, the term ‘Aβ plaques’ is increasingly common. For the following reasons, ‘Aβ plaques’ is recommended as the inclusive term that succinctly encompasses the multiplicity of these lesions under the umbrella of their shared feature - Aβ deposition:7 1) Aβ is present in all of the plaques that are linked to ‘normal’ aging and AD, regardless of size, shape, aggregation state, location, or overall composition. 2) The term ‘senile’ is vague and arbitrary, and not all plaques occur in ‘senile’ humans. Although Aβ plaques become more common at older ages, they can emerge in the 4th decade of life or earlier, especially in people with some autosomal dominant forms of AD [126].8 3) Plaques that are structurally similar to Aβ plaques occur in other neurodegenerative disorders, yet these result from the misfolding and aggregation of different proteins. Such plaque-forming proteins include the prion protein (PrP) in certain spongiform encephalopathies [127, 128], the ABri protein in Familial British Dementia [129, 130], and the ADan protein in Familial Danish Dementia [131, 132]. 4) Not all Aβ deposits incorporate abnormal neurites, which often are sparse or absent in diffuse plaques [133] including cotton-wool plaques [90, 93, 134] (see below). The term ‘neuritic plaques’ is suitable for the lesions that contain neurites, but these are only a subset of the entire family of Aβ plaques. 5) The Aβ in plaques does not always meet all of the criteria for amyloid [6] (see Section 1.2). Many diffuse Aβ deposits in the aging brain do not show birefringence after staining with Congo red. In addition, large, cotton-wool Aβ plaques lacking amyloid cores are abundant in certain presenilin-1 mutant cases of autosomal dominant AD [90, 91, 93, 94, 134] and in some non-familial cases [92]. (The Aβ in non-amyloid plaques from some presenilin-1 mutant cases is unusual in that it consists mostly of N-terminally truncated Aβ [94], as do diffuse deposits in the cerebellum in AD [135] and Down syndrome [135, 136]). The term ‘amyloid plaques’, like ‘neuritic plaques’, is appropriate for a subgroup of the lesions, but the universal constituent is Aβ, whether it is in the form of amyloid or not; hence, more precise designations of plaque subtypes would be, for example, ‘Aβ-amyloid plaques’ and ‘neuritic Aβ plaques’. Note that ‘diffuse plaques’ here refers to the fact that the Aβ accumulation is ‘widely spread or scattered; not concentrated’ [137], without consideration of the nature of the Aβ deposits, e.g., thread-like or punctate. ‘Diffuse’ thus denotes only the characteristics of the Aβ deposits, and not the dysmorphic neurites or any other component of the plaques. Also, when analyzing Aβ plaques histologically, it is useful to be cognizant of the plane of section, thickness of the tissue, and the limitations of a given staining protocol. Plaques are 3-dimensional structures that, when large enough, are only partially captured in thin histologic sections (Figure 4). Furthermore, different stains recognize different components of plaques. Consequently, a comprehensive assessment of plaques requires their full reconstruction and the application of suitable markers for potential components. In congruence with the trend to define AD according to its molecular underpinnings [109], defining the plaques that occur in aging and AD based on their principal proteinaceous component unambiguously distinguishes them from similar lesions in other disorders. In addition, this molecularly grounded moniker explicitly specifies the attribute that defines these plaques as unique pathologic entities: the misfolding and abnormal accumulation of the Aβ protein. 5 ‘plaque (Subject: Medicine and health): Any small patch or region of abnormal tissue within the body. See amyloid plaque, gliosis. [From French plaquer to plate, from Middle Dutch placken to beat metal].’ From: Oxford Dictionary of Word Origins: https://www.oxfordreference.com/view/10.1093/acref/9780199547920.001.0001/acref-9780199547920 6 Note that ‘Druse’ (‘geode’) differs from ‘Drüse’ (with Umlaut), which refers to a ‘gland’. 7 Because ‘Aβ’ and ‘plaque’ are both nouns, they could be connected by a hyphen (Aβ-plaque). I have chosen not to include the hyphen (the ‘open form’) in order to simplify usage. In some cases (such as Aβ-CAA & Aβ-oligomers), I have retained the hyphen for clarity. 8 W.H. McMenemey opined in 1963: ‘...the structures first observed by Blocq & Marinesco (1892) and thought by them to be nodules of glial sclerosis were called by Simchowicz (1910) ‘senile plaques’ - an unfortunate choice of name for it has coloured our thinking for the past fifty years’ [reference 509]. 4. The anatomic distribution of Aβ plaques 4.1 Histology Determination of the amino acid sequence of Aβ [2-4] prompted the development of sensitive and specific antibodies that have facilitated the investigation of the anatomic localization, structural diversity, and biochemical composition of Aβ deposits in the brain. Aβ plaques become increasingly frequent as age advances [80, 138], but they are especially numerous in AD patients. The anatomic distribution of Aβ plaques is variable, and it differs both among individuals and among brain regions in a given person [139-141] (Figure 5). In general, association areas of the neocortex are more vulnerable and/or affected earlier than are primary motor and sensory areas [140]. Aβ deposition is particularly profuse in the default mode network, an interconnected assemblage of brain regions that maintain vigorous metabolic activity when the brain is in an otherwise resting state [142]. The structure of Aβ plaques is influenced in part by the architectonic characteristics of the areas in which they form [139, 143], but it is usual for several kinds of plaque to intermix within a given site (Figure 6). In the neocortex, the laminar distribution of diverse Aβ plaques can vary markedly [140] (Figure 5).
Figure 5. Variation in Aβ deposition in adjacent cortical gyri from an AD patient. Antibody 4G8, Nissl counterstain. Bar = 500μm.
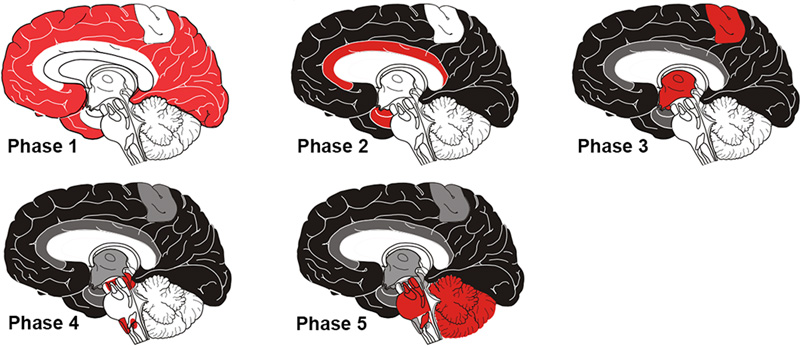
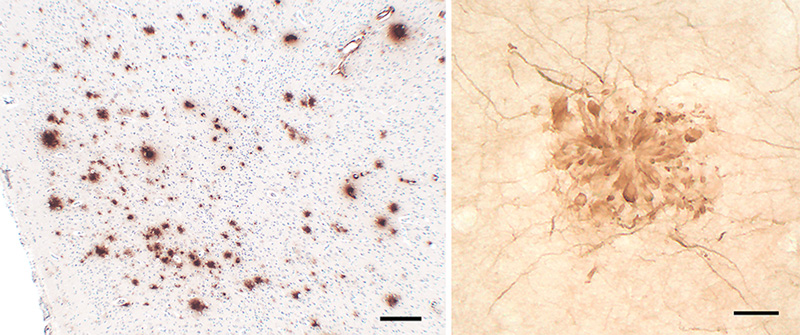
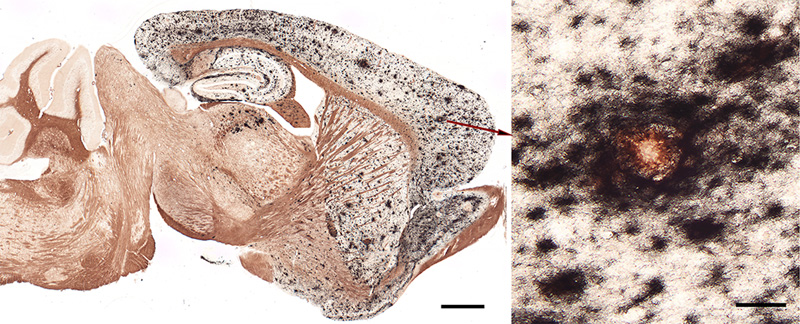
Figure 6. Variable morphology of Aβ plaques in the cortex of an AD patient. Classical dense-cored plaques with the core-space-corona pattern are in the upper left and lower right, and an irregular cloud of diffuse material is near the center, along with numerous very small patches. Antibody 4G8; Nissl counterstain. Bar = 50μm. Based on an analysis of human brains with different degrees of plaque accumulation, a spatiotemporal course of Aβ plaque formation has been proposed [19, 144, 145]. There is general agreement that diffuse plaques are the earliest type to emerge, followed later by cored (amyloid) plaques [146]. According to Thal and colleagues, in the first phase of the process, diffuse Aβ plaques appear in the neo(iso)cortex; in the second phase, allocortex, the hippocampal formation and amygdala are affected; in the third phase, plaques arise in the basal ganglia and diencephalon; in the fourth phase they appear in the midbrain and medulla oblongata; and in the fifth phase, the pons and cerebellum are affected [19, 144, 145] (Figure 7). These stages have been consolidated by Serrano-Pozo and colleagues [133] into an isocortical stage 1, allocortical/limbic stage 2, and subcortical stage 3. This general pattern of spread has been confirmed by a cross-sectional in vivo analysis of Aβ-amyloid deposition profiles using Florbetapir-PET imaging [147]. Thus, in the end-stage of AD, most brain areas exhibit at least some Aβ deposition. The spinal cord has been less studied; while it appears to be largely spared, plaques there have been reported in some instances [93, 148].
Figure 7. The phases of Aβ plaque distribution in the brain [references 19, 145]; illustration courtesy of Dietmar Thal, KU Leuven. 4.2. In vivo imaging At the turn of the 21st century, the first imaging agents were introduced to detect amyloid in the living human brain via positron emission tomography [149, 150]. Jorge Barrio and colleagues introduced 2-(1-[[6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl]]ethylidene) malononitrile ([18F]FDDNP), which binds to both Aβ-amyloid and tau tangles, and which has achieved some utility in diagnosing tauopathies [151-153]. A more Aβ-selective ligand, developed by William Klunk, Chester Mathis and colleagues, is 2-(4’-[11C]methylamino-phenyl)-6-hydroxybenzothiazole (Pittsburgh Compound-B [PiB]) [149, 154]. Derived from the chemical structure of the histologic staining agent Thioflavin-T, PiB crosses the blood-brain barrier and binds with high affinity and selectivity to Aβ deposits in plaques and CAA [154].9 PiB (which is labeled with carbon-11), was followed by similar PET ligands labeled with fluorine-18 (a radiolabel with a longer half-life than carbon-11): Florbetapir (Amy-vid) [155, 156], Florbetaben (Neuraceq) [157], and Flutemetamol (Vizamyl) [158]. By assessing Aβ-amyloid load in living subjects, these imaging agents have facilitated the differential diagnosis of AD and the longitudinal tracking of Aβ accumulation. They are particularly sensitive in detecting dense-core Aβ plaques, although they also bind to some extent to Aβ-CAA and diffuse Aβ deposits [47, 159-161]. Histochemical analysis of fluorescently labeled (‘clicked’) PiB applied to AD tissue sections confirms the preference of PiB at low concentration (100nM) for dense-core plaques [162]. Interestingly, PiB does not show significant high-affinity binding to Aβ-amyloid deposits in aged nonhuman primates with substantial Aβ burden [163], even though the amino acid sequence is identical to that of humans (see Section 10). (Note that binding of ligands can vary among humans; for example, a case of end-stage AD has been reported with extraordinarily high Aβ load, a predominance of Aβ40, and minimal high-affinity binding of PiB [164]). Since neither AD-like tauopathy nor dementia has been reported in nonhuman primates [165], comparative analysis of ligand binding could be useful in defining the variant molecular characteristics of Aβ deposits and their relationship to disease phenotype (see Sections 5.2 and 10). 9 In the 1920s, Congo red was introduced as an in vivo diagnostic agent for non-cerebral amyloidosis. Following intravenous injection, the rate at which Congo red was cleared from the blood was thought to reflect amyloid burden in affected organs (the more amyloid to bind the dye, the more rapid its clearance from blood). For various reasons, the test never achieved widespread use (see Buxbaum and Linke [2012] [reference 52]). 5. The variety of Aβ deposits 5.1 Aβ plaques The histologic implementation of specific antibodies in the 1980s firmly established that Aβ plaques in the brains of Alzheimer patients comprise a remarkable variety of morphologies [143, 166-172]. Several modern classification schemes have been proposed (e.g., [143, 166, 173-176]), and while there is not universal agreement on some of the terms, Aβ plaques can be broadly categorized into amyloid plaques per se (with dense, congophilic cores), and a range of more loosely organized deposits of myriad sizes, shapes, densities and locations [133] (Figures 5, 6, 8, 9). It is noteworthy that different genetic mutations can be associated with particular predominant plaque morphologies, as well as the presence of CAA (see Alzforum for a list of Alzheimer-associated mutations (https://www.alzforum.org/mutations). Note also that relatively few of the mutant forms of AD have been thoroughly scrutinized neuropathologically. Within the general categories of plaque structure, the Aβ-amyloid plaques are more or less spheroidal lesions that include ‘classical’ or ‘mature’ plaques and so-called ‘burned-out’ or ‘compact’ plaques [177, 178]. Recently, a ‘coarse-grain’ plaque type with multiple small cores and a predominance of Aβ40 has been described in advanced AD cases, often in association with APOE4 homozygosity and CAA [179]. Diffuse Aβ plaques are much more numerous than are amyloid plaques in the Alzheimeric brain [143] (Figures 6, 8), and they span a range of compactness from vaguely Aβ-immunoreactive, Congo red-negative regions (e.g., ‘fleecy’ plaques [180]) to clusters of loose fibrillar material that sometimes are weakly congophilic [139, 166]. Ultrastructurally, some of these diffuse deposits have been shown to include amyloid fibrils [181-183], whereas others do not [183], the latter possibly representing a pre-amyloid stage of Aβ aggregation [139].
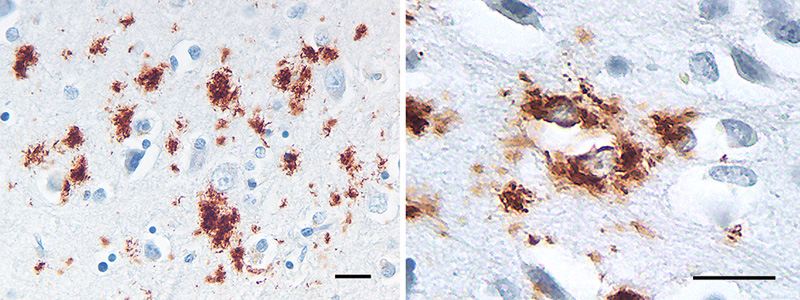
Figure 8. Small, often stellate Aβ deposits in the cortex of an AD patient. Some Aβ accumulates within glial cells, most likely astrocytes (right). Antibody 4G8; Nissl counterstain. Bars = 20μm.
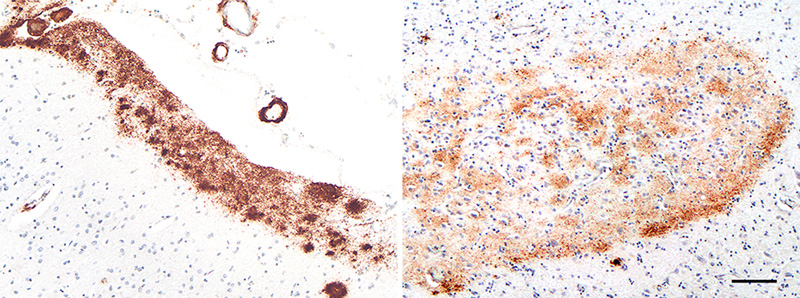
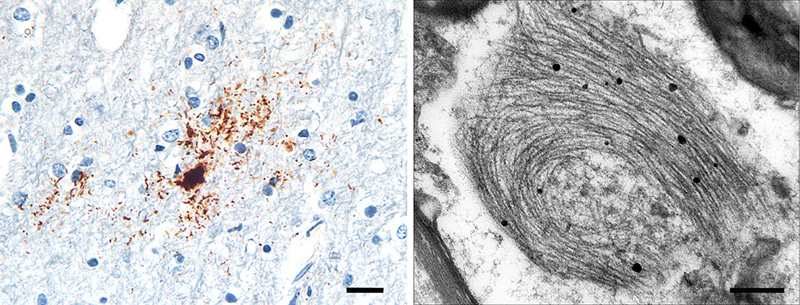
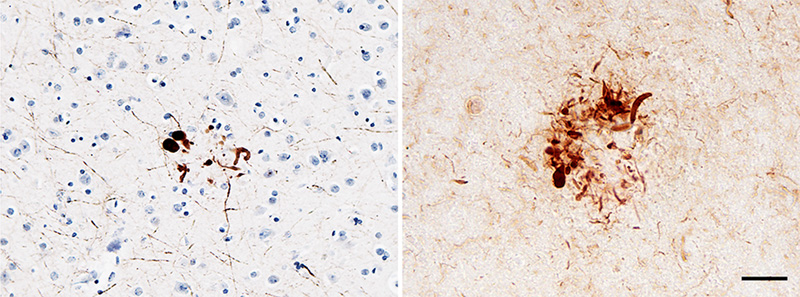
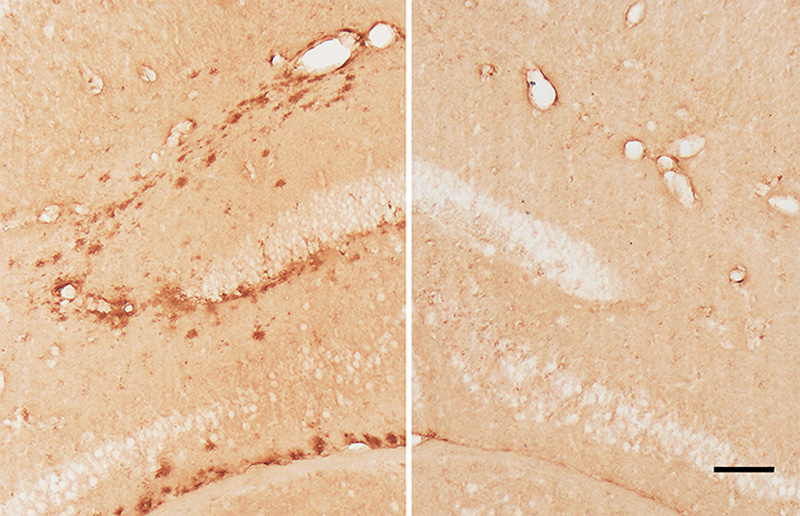
Figure 9. Band-like subpial Aβ (left) in neocortical layer 1 and presubicular lake-like Aβ (right) from two cases of AD. The subpial Aβ can be discontinuous, confluent, or punctate. Antibodies 4G8 (left) and 6E10 (right); Nissl counterstain. Bar = 100μm for both images. Diffuse Aβ plaques comprise very small, often stellate assemblies scattered about the parenchyma (Figure 8), a sheet-like band of sometimes confluent, sometimes patchy material in the subpial cortex (Figure 9), large ‘cotton-wool’ plaques, and very large ‘lake-like’ patches, including a distinctive cribriform deposit in the subicular complex [143, 171, 176, 184] (Figure 9). Abnormal neurites generally are absent or sparse in diffuse deposits [139], and this includes the cotton-wool plaques that are characteristic of some advanced AD cases [90-94, 134]. Despite their abundance in the Alzheimeric brain, very small diffuse deposits have received remarkably little scientific attention [175]. These probably correspond to the small (~2μm diameter) ‘Sternchen’ which Fischer in 1910 considered to be the first stage of plaque formation [185]. At least some of them appear to be related to astrocytes [175, 186] (Figure 8), but the absence of systematic research on these ubiquitous lesions currently precludes meaningful consideration of their involvement in the proteopathic process. Similarly, the immunoreactivity of some vestigial (extracellular) neurofibrillary tangles with antibodies to Aβ [187-194] (Figure 10) remains mechanistically undefined.
Figure 10. Neurofibrillary tangles in the cortex of an AD patient immunostained with an antibody to Aβ40. When present, this colocalization occurs mostly on extracellular ('ghost') tangles. Nissl counterstain. Bar = 50μm. Certain types of Aβ plaque are typical of the brain compartments in which they develop, e.g., in the cerebellum, basal ganglia, or different cortical regions and laminae (see [139]). In the white matter, distinctive granular accumulations of Aβ [143] occur to varying degrees (Figure 11). These clusters consist of fibrillar Aβ lying outside of the axons, and they appear not to be associated with obvious tauopathy or other abnormalities of the axons themselves [143], although their functional significance is largely unexplored. The core-space-corona arrangement of Aβ is a notable structural feature of classical Aβ plaques that was noted in several early investigations (reviewed in [116, 119, 120]). These subdivisions of plaques have been given various designations in the early literature, for instance Zentrum or Kern, Hof, Ring, etc.10 In tissue that has been immunostained for Aβ, classical Aβ plaques have a condensed core of Aβ-amyloid surrounded by an optically clear region with little Aβ, and then an outer corona of more diffuse Aβ [195] (see Figure 1); the relatively clear intermediate space and the outer corona are occupied by neuronal and glial elements (which are considered in more detail in Section 6).
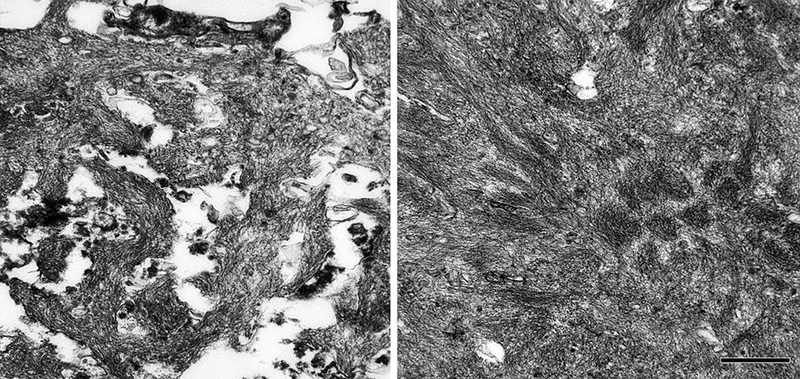
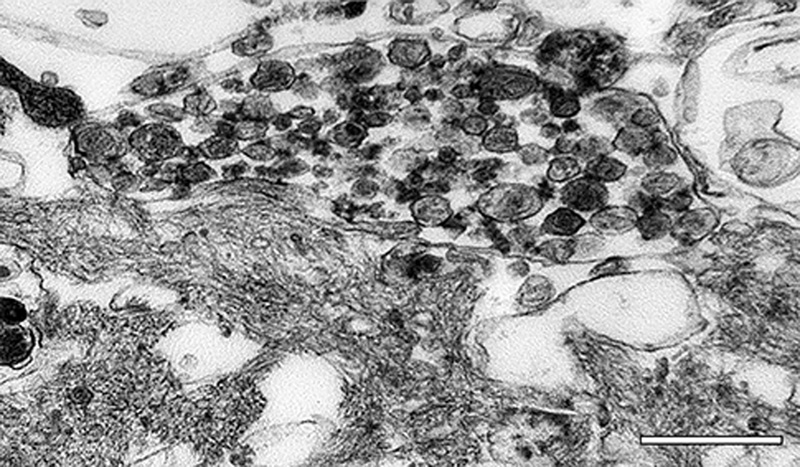
Figure 11. Aβ deposits in white matter of an AD patient comprise clusters of small puncta and filamentous bundles. Left: Light-micrograph of a cluster immunolabeled with antibody 4G8 (Nissl counterstain). Right, electron micrograph of a punctum immunolabeled with antibody 4G8 (black dots are gold particles bound to the secondary antibody). Bars = 20μm (left) and 200nm (right). Viewed in the electron microscope, Aβ-amyloid fibrils in the plaque core are densely packed and often bundled to form a patchy matrix, and viable cellular processes there are largely absent. The more loosely organized Aβ-amyloid sheaves in the space and corona interdigitate with cellular elements such as glial processes and neurites (Figure 12; see also Figures 18 and 20). Embedded in the fibrillar meshwork of amyloid in plaques, various small, spherical particles can be seen (Figure 13). The origin and significance of this material is obscure, but it could account for some of the non-Aβ substances that have been detected in the cores of Aβ plaques (see Section 7). One possibility is that these vesicles originate from intracellular multivesicular bodies, which have been shown experimentally to be an important site of APP/Aβ biology [196-201]. In this regard, vesicular structures ranging from 50 to 300nm in diameter have been reported among the amyloid fibrils in a cell culture model of Aβ amyloid deposition [202]. The center of the compact core in some Aβ-amyloid plaques is refractory to Aβ-immunostaining (Figure 14), even though it is positive for the amyloid-selective dyes Thioflavin-S and Congo red [203]. Ultrastructural analysis indicates that the material in the center of fully developed plaques often has a more granular, amorphous appearance (Figure 13) than the obvious fibrils in the mantle of the core and in the periphery. Classical Aβ-amyloid plaques are often ascribed special relevance to neurodegeneration [1, 204], as they are much more likely to involve neuritic malformation and reactive gliosis than are the diffuse deposits [133]. In this regard, it is noteworthy that cognitively normal elderly subjects with abundant Aβ plaques tend to have mostly diffuse plaques [1] with few neurites and little glial reactivity [139]. However, as noted above, there are rare cases of advanced AD in which classical plaques or dense-cored plaques are infrequent [90-93], suggesting that amyloid per se is not essential to the development of dementia. A similar situation holds for prion diseases, all of which are linked to the misfolding and self-assembly of PrP [205, 206]; in some prionoses (such as Gerstmann-Sträussler-Scheinker syndrome and new-variant Creutzfeldt-Jakob disease), PrP-amyloid plaques can be numerous, whereas in others, little if any amyloid is present [127]. In these instances, oligomeric species of the proteins may have particular importance [20], although this has not been definitely established.
Figure 12. Ultrastructure of fibrillar Aβ in the plaque corona (left) and core (right) in an AD patient. Bar = 500nm for both images. A small proportion of Aβ-amyloid plaques lack the outer corona and have few or no neurites; these relatively plain structures have been thought to represent an end-stage in the evolution of plaques, and so were dubbed ‘burned out’ plaques [143, 178]. Based on their apparent sequential appearance ance in the AD brain, a progression has been proposed in which plaques originate as diffuse (‘primitive’ or ‘immature’) deposits that evolve into classical (or ‘mature’) Aβ plaques and then finally into burned-out plaques [143].11 While longitudinal studies in mouse models of cerebral Aβ accumulation have begun to shed light on the time-course of plaque development (see Section 10.2), the order of events in the human brain is still speculative [207].
Figure 13. High-magnification electron micrograph of a portion of the core of an Aβ-amyloid plaque in an AD patient. The fibrillarity of the material is less evident than in more peripheral zones. Unidentified particles (2 are marked by arrows) of various sizes and densities are interspersed among the amyloid fibrils; these can be found both in the core and corona. Bar = 200nm.
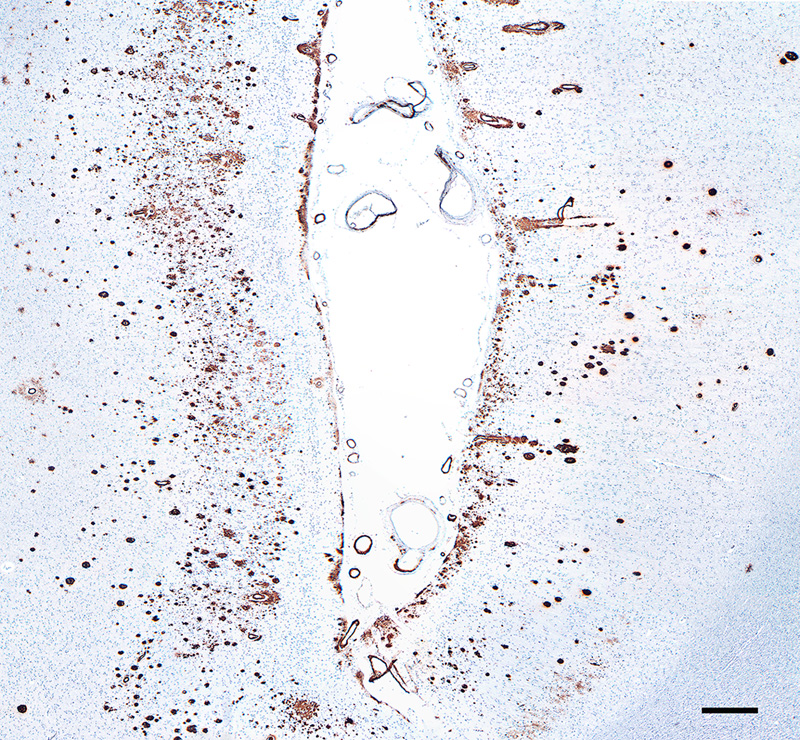
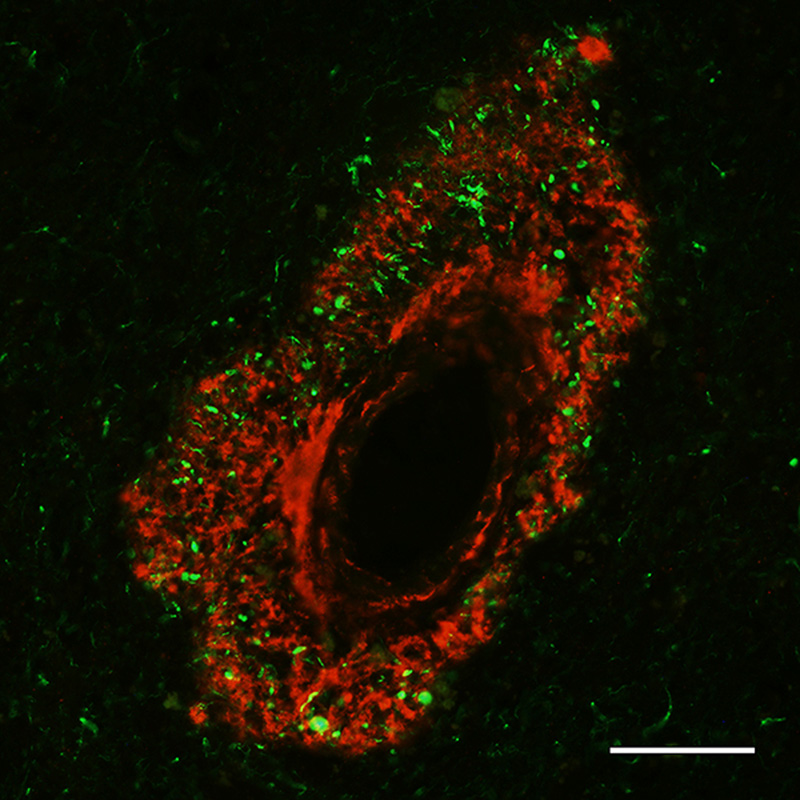
Figure 14. Aβ plaque with an antibody-refractory central core in an AD patient. Antibody 6E10; Nissl counterstain. Bar = 20μm. Biochemical determination of the age of Aβ deposits indicates that the amyloid core is older than the diffuse Aβ in the corona and in diffuse plaques [208, 209]. Armstrong [173] has suggested that the major plaque types mostly arise independently, rather than in an evolutionary progression. In any case, the transformation of diffuse plaques into compact amyloid might not be an inevitable occurrence; for instance, it appears that diffuse Aβ deposits such as the lake-like cloud of Aβ in the subicular complex (Figure 9) do not progress into dense masses of amyloid, and this may be true also for cotton-wool plaques in AD cases with certain presenilin-1 mutations [90, 91, 93]. Finally, it should be emphasized that the relative pathogenicity of the many different Aβ plaque types in the aging human brain remains ambiguous. It is fairly certain that reactive gliosis/inflammation and the local disruption of neuronal processes in classical Aβ plaques are deleterious to brain function (see Section 6), but it is likely that oligomeric agents are the more directly injurious manifestation of misfolded proteins (see Section 1.1). In fact, while the plaques themselves are indicative of a pathogenic molecular process, in and of themselves they may be relatively benign or even protective [210, 211], at least when inflammation and surrounding oligomers are negligible (see Sections 1.1 and 6). 10 Fischer [1910] [reference 185] referred to the central core as the ‘Morgenstern’ (morning star), and described the structure of one type of plaque thusly: ‘Auch hier ist ein zentraler Morgenstern, aus dem mehr oder weniger lange Büschel entspringen; der Fädchenring ist ziemlich weit vom Zentrum entfernt, so dass ein grosser Hof entsteht, der von den Strähnchen durchzogen wird’ 11 Diffuse plaques have long been considered an early stage in plaque formation (see, e.g., Critchley [1929] [reference 116]). 5.2 Aβ strains In AD, the diverse morphological attributes of plaques might reflect, in addition to the local tissue organization, the variable truncation, folding, and molecular architecture of Aβ [212, 213]. These variants are referred to as proteopathic strains, a biological concept that was adopted by the spongiform encephalopathy community to explain the alternative disease phenotypes resulting from prion infections [214, 215]. At the molecular level, the formation and propagation of Aβ aggregates (as well as the proteins involved in several other proteopathies [216]), constitute a mechanism that is fundamentally similar to that of prions [217, 218] (see Section 9). The capacity to spawn distinct strains is considered to be a shared property of proteins that are prone to misfolding and self-assembly [56, 59, 219]. In vitro, a given protein can create morphologically diverse amyloid fibrils under different environmental influences, for example temperature, pH, ionic strength, protein concentration [220, 221] and agitation [67, 222]. Strain properties can be conveyed to newly forming amyloid fibrils; in vivo, it is thought that proteopathic strains undergo conformational selection by which the strain best suited to a given environment predominates [215, 220, 223, 224]. Studies in genetically modified mouse models (which can be customized to make various types of Aβ) can shed light on the factors that govern alternative plaque morphologies in the living brain [225]. The generation of Aβ strains is influenced by characteristics of the aggregating Aβ such as mutations, truncations and chemical modifications (see Sections 1 and 7). Aβ forms distinct structural strains in different subtypes of AD [226-231]. Investigations of the molecular configuration of Aβ fibrils in vitro have yielded insights into potential determinants of Aβ strains (see, e.g., [228, 232-235]), but cryo-electron microscopic analysis of meningovascular Aβ-amyloid indicates that Aβ-CAA fibrils formed in vivo, though polymorphic, differ in important ways from those formed in vitro [66]. A similar analytic comparison of Aβ fibrils from plaques in the brain parenchyma and CAA could help to explain the inconsistent co-presence of plaques and amyloid angiopathy in AD. 5.3 Cerebral Aβ-amyloid angiopathy (Aβ-CAA) Several different proteins can form CAA in different disorders, but Aβ is the most common source of CAA in the elderly [236]. Aβ accumulates in the vascular wall and perivascular zone in cases of primary Aβ-CAA involving mutations in the gene for APP [21, 237-240] and - to varying degrees - in nearly all cases of AD [241-244]. AD and Aβ-CAA share many genetic risk factors, and like Aβ plaques, idiopathic Aβ-CAA sometimes is present in the nondemented elderly [240, 241]. CAA is a significant risk factor for lobar hemorrhage [236, 245], particularly in individuals with hypertension [246]. In end-stage AD, the amount of Aβ-CAA varies widely, even in the presence of copious plaques [247], although the severity of Aβ-CAA tends to increase with increasing plaque load [21]. Furthermore, in some instances, Aβ-CAA can emerge in the absence or near absence of Aβ plaques, notably in an autosomal dominant form of Aβ-CAA known as hereditary cerebral hemorrhage with amyloidosis (Dutch type) (HCHWA-D) [239, 248, 249]. There is evidence for some diffuse parenchymal Aβ deposition [250, 251] and cognitive decline [238, 252] in these cases, but the clinical phenotype probably reflects the vascular pathology more than an AD-like disorder in which plaques and tangles are abundant [253]. Cognitive dysfunction [254-258] and neurodegenerative changes [259] also have been associated with idiopathic Aβ-CAA. In approximately 25% of end-stage AD patients, Aβ-CAA affecting large vessels is considered to be severe (arterioles are more often afflicted than are veins); capillary Aβ-CAA is less common, being severe in approximately 10% of cases [247]. In advanced Aβ-CAA, the amyloid often extends through the tunica adventitia and into the surrounding parenchyma, where it is pervaded by tau-immunoreactive abnormal neurites [25, 260, 261] (Figure 15). For unknown reasons, in regions of the neocortex where capillary Aβ-CAA is focally abundant, Aβ plaques are relatively scarce [25, 247, 262].
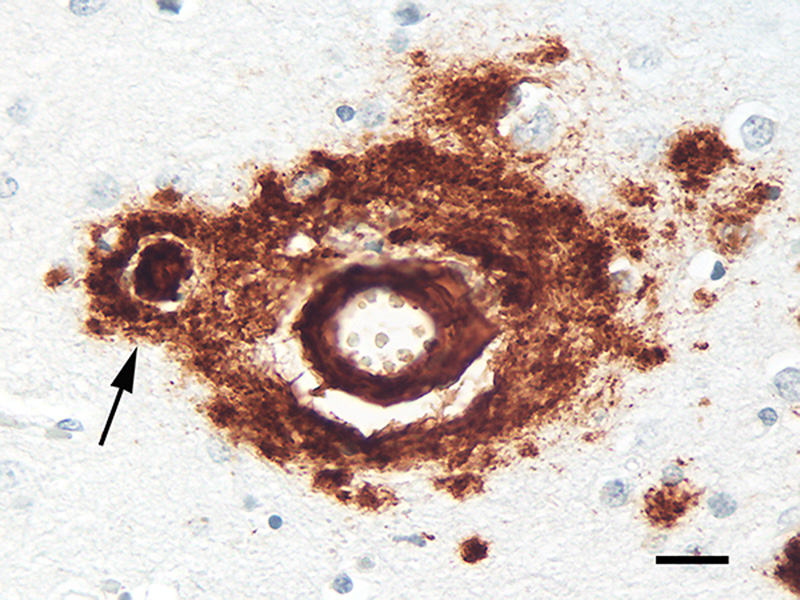
Figure 15. Fluorescence-immunolabeled dyshoric cerebral Aβ-amyloid angiopathy (red; antibody R398) and tau-immunoreactive neurites (green; antibody CP13) in the cortex of an AD patient. Bar = 50μm. In the early stages of large-vessel Aβ-CAA, Aβ42 is more commonly present than is Aβ40 [263], but in later stages Aβ40 predominates [263, 264]. Capillary Aβ-CAA, however, more often is positive for Aβ42 than for Aβ40 [263]. It has been suggested that the deposition of Aβ in capillaries transpires by a different mechanism than that in large vessels and Aβ plaques [25, 26]. Quantitative spatial analysis has largely refuted the hypothesis that cerebral capillaries are the nidus of Aβ plaque formation [265]. Interestingly, ‘coarse-grain’ plaques, a special type of lesion (see Section 5.1), are more common in cases with abundant Aβ-CAA, particularly capillary Aβ-CAA [179]. Aβ-CAA, like Aβ-plaques, is associated with reactive gliosis and a perivascular inflammatory response [240, 260], although the presence of frank perivascular inflammation is inconsistent [25, 266]. Aβ-amyloid plaques are occasionally confluent with Aβ-CAA (‘juxtavascular plaques’; Figure 16), but the etiologic relationship between these merged lesions is uncertain.
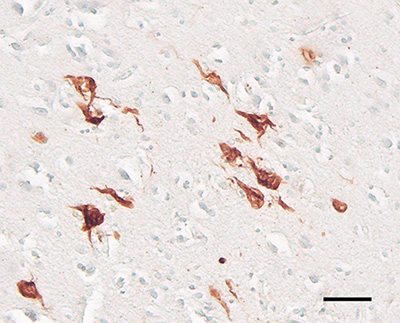
Figure 16. Juxtavascular Aβ-plaque (arrow) in the cortex of an AD patient. Antibody 4G8, Nissl counterstain. Bar = 20μm. Various genetic, biochemical and pathophysiologic factors appear to influence how the misfolding and aggregation of the same protein - Aβ - can lead to two different phenotypic presentations - parenchymal plaques and vascular amyloid [21]. While many auxiliary molecules are present in both Aβ plaques and Aβ-CAA, some are not shared by the two lesions [267]. Thus, Aβ-CAA and Aβ plaques likely result from at least partially distinct ontogenetic pathways [21] (in this regard, it is noteworthy that the disappearance of plaques in Alzheimer patients immunized against Aβ is accompanied by a [possibly transient] increase in Aβ-CAA, suggesting a transfer of Aβ from the parenchyma to the walls of blood vessels [268]). For in depth reviews of CAA, see [21, 236, 237, 240, 241, 260, 269]. 6. Cellular components of Aβ plaques The main cellular elements - neuronal processes and glial cells - in classical plaques were well-documented by pioneering investigators in the field (see [116]),12 although the nature of their involvement, and their functional relationship to the core, have been a persistent matter of speculation [207]. Diffuse deposits of Aβ mostly lack obvious changes in local neurons and glial cells, whereas these cells are conspicuously altered in classical Aβ plaques. Since classical plaques are especially numerous in most cases of late-stage AD, the associated abnormal neurites and activated glial cells probably contribute to the disturbance of brain function by the plaques [133]. 12 Early descriptions of plaques included drawings that enabled the artist to clearly depict all cellular elements throughout the depth of the tissue sections in a way that photomicrography, still in its infancy, could not. The result was sometimes striking images that have been difficult to surpass in the century-plus since (see, e.g., the fine reproductions in DeFelipe [2010] [reference 510]). 6.1. Abnormal neurites In advanced AD, many Aβ plaques are decorated with an impressive profusion of dysmorphic neurites (Figures 1, 4, 17). Both axons and dendrites contribute neurites to plaques [207, 270]. Although most swollen neurites have been reported to be axonal in origin [138, 178, 207], a quantitative analysis of plaques in humans using axon- and dendrite-specific markers is needed to establish the relative involvement of these neuronal processes. Tortuous, atypical neurites that are not spatially associated with plaques are fairly common in the aging brain [139], but neuritic pathology is particularly evident in many Aβ-amyloid plaques. By disrupting the structure and trajectory of neuronal processes, Aβ plaques are thought to interfere with the connectivity and network functionality of the brain [38].
Figure 17. Abnormal neurites associated with cortical Aβ plaques in two AD patients. Left: immunostain for neurofilament-H (antibody SMI31) with a Nissl counterstain; right, immunostain for a conformational epitope on tau filaments (antibody MC1). The presence of aberrant neurites that are immunoreactive for these antigens in plaques is variable. Bar = 25μm (right) and 50μm (left). Abnormal neurites are heterogeneous in size, shape and content. Ultrastructurally, plaque-associated neurites may contain any of a number of inclusions, including, in addition to paired helical filaments, profuse mitochondria, various dense bodies, membranes, and multivesicular profiles [139, 178] (Figure 18). The mitochondria appear to be in different stages of degeneration, and they have been hypothesized to be a source of the Aβ-amyloid in plaques [207], as have multivesicular bodies [199, 271, 272]. The cytoskeleton is disrupted in swollen neurites [273], and studies of mouse models found that neuritic calcium (Ca2+) homeostasis [274] and autophagy [275] are dysregulated in them. Dickson [139] divided abnormal neurites into paired helical filament (PHF)-type neurites, which are characteristic of advanced AD, and dystrophic-type neurites, which are relatively more frequent in the plaques found in aged, non-demented subjects (and in animal models, in which PHFs per se are rare or absent [165]; see Section 10). Dickson also notes, however, that many neurites have the properties of both types, and that abnormal neurites tend to arise from axons or dendrites that just happen to be in the vicinity of the plaque [139]. This is likely to be a general rule for the presence of specific types of neurites in plaques, including those containing markers for diverse neurotransmitters (see below) and, e.g., the alpha-synuclein-positive neurites in Aβ plaques that are sometimes co-morbid with synucleinopathy in Lewy body disease [276].
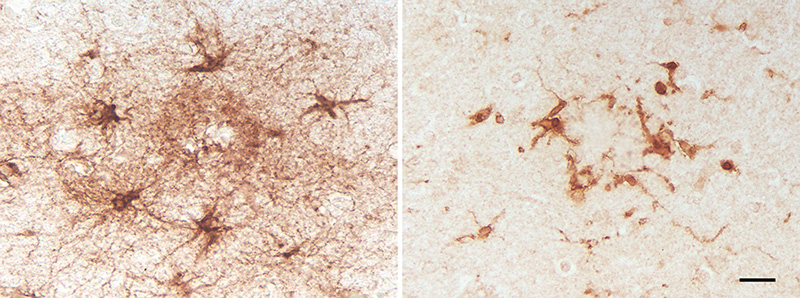
Figure 18. Abnormal neurite (top) containing organelles /debris adjacent to fibrillar amyloid (bottom) in the plaque corona of a patient with AD. Bar = 500nm. Histochemically, lysosomal enzyme activity is pronounced in dystrophic neurites, as is histochemical reactivity for APP and markers of degeneration such as chromogranin-A and ubiquitin [139]. The chemical variability of neurites may reflect, in addition to the neurons of origin, their stage of development and their response to injury or stress [277]. Several early researchers, including Fischer [117] and Ramon y Cajal (see [278]), thought that the swollen neurites in plaques represented an attempt by the neuronal processes to sprout. Since then, multiple growth-promoting factors have been detected in these neurites [278, 279], and Aβ deposits have been shown experimentally to induce axonal sprouting in the mouse brain [280]. Considered as a whole, these observations indicate the presence of both degenerative and regenerative mechanisms in the aberrant neuronal processes that are associated with Aβ plaques [133, 279]. Analyses of Aβ plaques in humans and aged nonhuman primates found that many different neurotransmitter systems contribute anomalous neurites to plaques [281-287], and that an individual plaque can contain neurites from multiple sources [288, 289]. These studies cast doubt on the hypothesis [290] that plaques emerge from the regression of neurites from a specific transmitter system, in particular the acetylcholinergic neurons of the basal forebrain [141]. Rather, they highlight the probable role of a common catalyst (e.g., misfolded Aβ and/or reactive glia) in driving neuritic dystrophy [139, 289, 291]. Indeed, the influential model proposed by Wisniewski and Terry [178] (see also [81]) that posited neuritic abnormalities in general as the initial stage of plaque ontogeny now seems untenable, especially in light of genetic findings implicating Aβ as the prime mover in the pathogenesis of AD [9, 22, 44, 45, 212]. Even so, dysmorphic neurites do influence the pathologic plaque milieu [207], and it is possible that, by releasing Aβ into the extracellular space, they contribute to the growth of plaques [272]. In addition, neuritic Aβ plaques are generally more strongly associated with dementia than are diffuse plaques [1, 133, 204]. Finally, the loss of synapses correlates strongly with the degree of dementia in AD [292-295]; synaptic pathology is especially evident in the immediate vicinity of Aβ plaques (see [296, 297], possibly owing to increased oligomeric Aβ in this region [297]. 6.2 Glial cells Of the many genetic risk factors for AD [298], two of the most potent variant genes - APOE (apolipoprotein E) and TREM2 (triggering receptor expressed on myeloid cells-2) - are highly expressed in glial cells [115], as are several other AD-associated genes [299-301]. Astrocytes and microglia are protean and interactive components of the homeostatic intrinsic immune system in the brain and spinal cord [299, 302, 303]. Histologic, genetic, biochemical and physiological findings strongly implicate them in the pathogenesis of AD [111-115, 299, 303-310] (Figure 19). Microglia and astrocytes do not operate independently of one another, but rather jointly influence Aβ processing and plaque biology [311, 312]. In the vicinity of Aβ-amyloid, these glial cells together form a partially integrated ‘reactive glial net’ [313] that, while considered to be an attempt to shield nearby neurons from Aβ aggregates [314], ultimately engenders a neurotoxic inflammatory microenvironment [313].
Figure 19. Reactive astrocytes (left; antibody to GFAP) and microglia (right; antibody to IBA1) in cortical Aβ plaques of two AD patients. Despite some overlap of the two cell types within plaques, astrocytic somata tend to be more peripherally located than are microglial somata. Bar = 20μm for both panels. Inflammation is both a risk factor for, and a result of, the deposition of Aβ in the brain [45, 315]. The recruitment and activation of glial cells by Aβ plaques has been likened to a local inflammatory reaction to a foreign body [138, 139], although glia contribute to the pathobiology of plaques in complex ways [299, 302, 305, 309, 312, 316-318]. Mouse models have enabled a dynamic view of glial function and the general biology of plaques, whereas the genetic and physiologic analysis of glia in human AD is much less advanced [299]. Even given the caveat that glia differ in humans compared to other species [316, 319, 320], mice have furnished unique insights into glial functionality in the living brain [304, 316, 321-324]. A growing literature underscores the ability of both microglia and astrocytes to adopt different physiologic states that influence how they contribute - positively or negatively - to AD (see, e.g., [299, 303, 306]). Current views of glial cells thus emphasize their dual role in the pathobiology of AD: they participate in the clearance of aberrant Aβ and other debris, but they also can secrete a variety of inflammation- and cell-stress-related molecules [304, 325, 326]. Much contemporary research seeks to define and disentangle these intricate and seemingly incompatible mechanisms. 6.2.1 Microglia Activated microglia are intimately associated with the fibrillar Aβ in classical Aβ plaques [139, 327-330] (Figure 20). They occupy much of the space between the plaque core and outer corona, and their processes interdigitate with the bundles of amyloid [311, 327]. The discovery that loss of function mutations in TREM2 are a strong risk factor for AD has heightened interest in the role of microglia in neurodegeneration [299, 306]. TREM2 is a cell-surface immune receptor on many myeloid cells, including microglia, which exclusively express TREM2 in the brain [306]. The production of TREM2 is increased in AD [331], and it mediates the activation and responsiveness of microglia to Aβ-amyloid plaques [332]. Microglia have been thought to either phagocytose [139] or produce [311] multimeric Aβ, and their functional variability makes both actions conceivable, depending on the circumstances. On the one hand, there is evidence that microglia normally impede the generation of Aβ plaques; inhibition of microglial functionality in mice was found to increase plaque load [333, 334], and microglia contribute to the clearance of dense-core plaques following anti-Aβ immunization therapy [335] (see also the analysis of immunized humans by Nicoll and colleagues [336]). Additionally, studies in mice indicate that TREM2 signaling transforms homeostatic microglia into disease-associated microglia (DAM), in which state they phagocytose Aβ in plaques [306, 337]. Impeding TREM2 functionality in microglia reduces the binding of ApoE to Aβ-amyloid in plaques and augments the seeded propagation of Aβ-amyloid [338]. (Genetic knockout of TREM2 also promotes the seeded aggregation and spread of tau in neuritic Aβ plaques [339]). On the other hand, ultrastructural [311, 340, 341] and experimental [342] investigations have suggested that microglia can generate Aβ-amyloid fibrils. In support of this hypothesis, sustained pharmacologic reduction of microglia significantly diminished Aβ plaque load in a transgenic mouse model [343]. The ability of microglia to assume multiple phenotypic states underscores the complexity of their participation in the biology of Aβ plaques [299, 300, 305, 344-346]; they contribute to normal brain homeostasis, but they also have injurious properties, particularly when activated [299, 300]. In mice, microglia have been found to exhibit a range of activation states, each of which involves the expression of distinct gene modules [299]. Microglia become activated in the presence of aggregated Aβ, and in this condition they can harm the brain both through the secretion of pro-inflammatory agents and the elimination of synapses [300]. To complicate matters further, a variety of microglial phenotypes are simultaneously present within the same brain [345], and the involvement of microglia in plaques differs as a function of age and disease stage [299]. Finally, while the discovery of microglial risk factors for AD emerged from human genetic analyses [306], we know far more about microglia in rodent models than in human AD, and current evidence suggests that there are important differences that cannot be overlooked [299, 347-349]. These findings collectively highlight the challenges presented by microglia as therapeutic targets in AD.
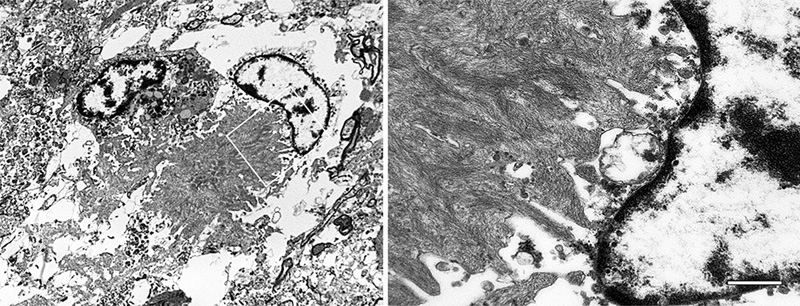
Figure 20. Electron micrographs of a microglial cell in an Aβ-amyloid plaque of an AD patient. The white box in the image on the left denotes the region at higher magnification on the right. The fibrillar bundles of Aβ interdigitate with the microglial soma. Note that the microglial cytoplasm appears artifactually rarefied in this autopsy-derived tissue. Bar = 500 nm (right), 2.8μm (left). 6.2.2 Astrocytes In the vicinity of many Aβ-amyloid plaques, astrocytes hypertrophy and increase their expression of glial fibrillary acidic protein (GFAP) [316] (Figure 19). The degree of astrocytic hypertrophy surrounding plaques, however, is inconsistent [317]. GFAP expression is a reasonably reliable index of astrocytic reactivity, but GFAP is not detectable in many healthy astrocytes, and its expression varies in different parts of the brain, in different animal species, and as a function of age.13 Compared to microglia, astrocytic somata tend to localize more peripherally to the aggregated Aβ in plaques [175, 302, 311, 313, 327, 330], whence their processes penetrate and to some extent encapsulate the plaques (Figure 19). Despite their tendency to partially segregate, astrocytes and microglia show some spatial overlap, and physical and chemical interactions between them help to define the inflammatory state of plaques [304]. Activated astrocytes promote the inflammatory milieu around plaques through the generation of pro-inflammatory substances, including cytokines/chemokines, activation of the complement cascade, and reactive nitrogen and oxygen species [316]. As in the case of microglia, the role of astrocytes in neurodegeneration is complicated by their variable and sometimes paradoxical phenotypes [317]. In AD, astrocytes can both gain a toxic function and lose their normal physiologic function [316, 350, 351]. Astrocytes have been shown experimentally to take up and degrade Aβ [315]. They also are capable of expressing Aβ [352], and astrocytes containing ample Aβ are present in the Alzheimeric brain [186, 316, 353, 354] (Figure 8). In addition, the extent of peri-plaque reactive astrocytosis is positively correlated with cognitive status in aged subjects, and their abundance is reduced in persons expressing apolipoprotein E4, a major risk factor for AD [316]. In summary, research on microglia and astrocytes has disclosed the extraordinary malleability of these glial cells, the complexity of their involvement in plaques, and thus the attendant difficulties in targeting them therapeutically. Interventions that modulate the activity of glia could either promote or hinder disease progression, depending on the state of the cells in different brain areas, their relative abundance, and the timing of therapeutic delivery in the protracted course of AD. Nevertheless, the obvious importance of microglia and astrocytes in the pathobiology of AD justifies continued efforts to decipher the mechanisms by which they interact with Aβ and with the other cellular components of plaques. For additional reviews of microglia and astrocytes in aging and AD, see [312, 355-357]. 13 The authors note that the findings should be interpreted cautiously in light of the pitfalls associated with histochemical methods (Garwood et al. [2017] [reference 316]). This advice applies to histologic analyses in general, as methods and interpretations can vary among laboratories (e.g., Alafuzoff et al. [2008] [reference 511]). 6.2.3 Oligodendrocytes Compared to microglia and astrocytes, oligodendrocytes have been less studied in AD [358]. Their involvement in plaques has long been debated (see, e.g., the contrasting views of Critchley [116] and Ferraro [118]: ‘Oligodendroglia apparently does not participate in the structure of plaques’ [Critchley, 1929]; ‘It is certain, then, that both oligodendroglia and microglia cells are usual components of senile plaques’ [Ferraro, 1931]). Soniat contended that oligodendrocytes are not integral to the formation of plaques, but rather, when present, their presence is purely coincidental [119]. A recent analysis, however, has revealed oligodendrocyte progenitor cells in Aβ plaques that become senescent and pro-inflammatory, in which state they are thought to augment the pathogenicity of aberrant Aβ [359]. More work on oligodendrocytes in association with Aβ plaques is clearly needed. 7. The broader biochemistry of Aβ in plaques The number of molecules that have been linked in some way to Aβ plaques is considerable (see, e.g., [139, 175, 278, 279, 360-364]), creating fertile ground for hypotheses on both the origin of plaques and the nature of AD. Along with the many substances directly associated with neurons and glia, aggregated Aβ itself is rich in accompanying molecules. Amyloid P component is present in different types of amyloid throughout the body [6, 365], including Aβ plaques [364, 366, 367]. Other molecules that have been reported to directly co-localize with at least some Aβ deposits include proteoglycans [6, 364, 368, 369], complement proteins [370-373], apolipoprotein E [374, 375], alpha-1 antichymotrypsin [376] and advanced glycation end products [377, 378], along with lipids, metal ions, reactive oxygen species and nucleic acids (see Stewart and Radford [364]). How Aβ-linked substances might be involved in the pathobiology of plaques is attracting increasing attention. For instance, a study in mice found that Aβ bound to nucleic acids acts as an immune signal, stimulating an antiviral response in microglia and astrocytes that instigates the complement-mediated elimination of local synapses [379]. The plaque-associated proteome can be interrogated by laser-microdissection of Aβ plaques followed by mass-spectrometric analysis [380-385]. These studies have identified numerous proteins that are enriched in plaques, though whether they are directly associated with multimeric Aβ or with the cellular constituents is sometimes undefined. It has been proposed that plaques mature through three biochemical stages within which the toxicity of the aggregates may differ; in stage 1, the aggregates lack both pyroglutamation at residue 3 (AβNp3E) and phosphorylation at residue 8 (pSer8Aβ); in stage 2, AβNp3E appears, and in stage 3, both AβNp3E and pSer8Aβ are present [19, 386]. Post-translational chemical modifications of Aβ can influence the aggregation of the protein along with the type of deposit that is formed in the brain [19, 387-391], but the mechanisms are, in many cases, still uncertain. 8. Microbes and plaques The notion that microbes might participate in the genesis of plaques has been considered at least since the early 20th century.14 Fischer likened mature plaques to actinomyces ‘Drusen’, although he noted that they were negative for multiple bacterial stains [117]. Critchley remarked in 1929 that the microbial origin hypothesis had failed to gain traction [116]. Despite more recent claims that senile plaques in AD ‘are made up by spirochetes’ [392], there is still no credible evidence that Aβ plaques are primarily collections of microbes or their remains. That said, there is fairly compelling evidence that certain microbial infections are risk factors for AD [393-395]. Perhaps the best evidence indicates that some herpesviridae increase the probability of developing AD [394, 396, 397]. Over 15 different microbes have been linked to AD by various researchers [398], but in many instances the findings are weak or contradictory (see, e.g., [399, 400]). In addition, it is important to distinguish cases of dementia in general (for which there are over 50 different causes [330]) from cases of dementia specifically due to the pathology of AD (as defined by Jack and colleagues [109]). It is fair to say that no known infectious agent is universally and exclusively associated with AD [395], but it seems likely that any of several types of brain infection (including chronic infection and/or reactivation of resident microbes) can accelerate plaque formation and the pathogenesis of AD [394, 395, 401, 402]. In other words, at least in some instances the development of plaques may represent a non-specific response to various infectious organisms. Aggregated Aβ has antimicrobial properties [395, 403, 404], and some microbial antigens have been reported in Aβ plaques [392, 405], but a systematic and comprehensive survey of microbial markers in different types of plaques and Aβ-CAA throughout the central nervous system has not been reported. APP-transgenic mice raised in a germ-free environment develop some Aβ plaques as they age, albeit fewer than mice raised in normal caging [406]. With the caveat that the mice strongly overexpress transgenic Aβ, the findings suggest that infection is not required for plaque formation, but that it can trigger and/or accelerate the process. The role of infection in the causation of Aβ plaques and as a risk factor for AD is an intriguing topic with potential implications for prevention and therapy, but supporting evidence for a specific role of specific microbes in pathogenesis is needed. For a critical consideration of the state of the field, see [393]. 14 I use the term ‘microbe’ here to include both conventional (living) microorganisms and viruses (but not prions). 9. The seeded induction of Aβ plaque formation The prion paradigm has become the dominant mechanistic explanation for the aberrant self-assembly and propagation of misfolded proteins in the brain and elsewhere in the body [58, 205, 218, 407-409]. At the molecular level, the prion paradigm postulates that misfolded, β-sheet-rich proteins aggregate into oligomeric/polymeric assemblies that can induce protein molecules of the same type to adopt a similar conformation. In this condition, the proteins tend to stick together, with the assemblies often (but not always) amassing into amyloid deposits. In the prion diseases, misfolded PrP self-assembles into highly stable multimers that are transmissible from one organism to another - the first verified instance of an infectious protein particle (‘prion’) [410, 411]. Human prion diseases also originate spontaneously or as a result of mutations in the gene for PrP [412]. The pathological signature of the prion diseases varies considerably [127, 128], but, as in AD, the universal feature of prionopathies is the accumulation of an abnormally folded protein - in this case PrP - in the nervous system. Systematic studies in transgenic mouse models expressing human APP have determined that Aβ plaque formation is driven by a molecular process that is indistinguishable from the mechanism by which prions instigate disease [217, 218, 413-415] (Figure 21). In this paradigm, brain extracts containing aggregated Aβ are infused into the brains of susceptible mice, instigating Aβ plaque development in a model-, dose- and time-dependent fashion [218, 407]. Analyses of seeded aggregation in experimental systems have demonstrated that Aβ seeds share key properties with prions: 1) they are protein-only agents that are resistant to destruction by heat and formaldehyde; 2) they incite the formation of cerebral Aβ plaques and Aβ-amyloid angiopathy when introduced into the brain or into the periphery; 3) they exist in multiple sizes; and 4) they can fold into different molecular variants referred to as proteopathic strains [212, 213, 217, 218, 407] (see Section 5.2). The strain-like properties of Aβ deriving from different subtypes of AD can be partially transmitted to plaques via exogenous seeding in mouse models [227, 230].
Figure 21. Seeded Aβ deposition in the hippocampal formation of a TG2576 APP-transgenic mouse 5-months following unilateral injection of dilute AD brain extract into one hemisphere (left). The contralateral hippocampus in the same tissue section is on the right. Antibody 4G8; Bar = 100μm. These investigations highlight the prion-like seeded aggregation of Aβ as the propulsive mechanism behind the formation of Aβ plaques. Since there is currently no evidence that AD or other cerebral proteopathies are infectious under ordinary circumstances [416, 417], it is likely that plaques ordinarily arise endogenously with the stochastic emergence, persistence and spread of Aβ seeds. This process can be advanced by various environmental and endogenous risk factors that influence the likelihood that Aβ will misfold and propagate in the brain [45]. Under extraordinary circumstances, however, Aβ plaques and Aβ-CAA can be instigated by exogenous Aβ seeds in humans [417]. Treatment of young people with growth hormone derived from cadaveric human pituitary glands, beginning in the late 1950s, resulted unexpectedly in the development of prion disease (Creutzfeldt-Jakob disease) many years later [418, 419]. The apparent cause was the presence of infectious prions in the preparations, probably because the large batches of pituitaries that were homogenized for extraction of growth hormone contained some glands from decedents with prion disease. Researchers in England later found that both Aβ plaques and Aβ-CAA were much more common in human growth hormone-treated subjects than in non-treated controls [420]. Furthermore, Aβ deposition was precipitated both in growth hormone recipients dying with [420] or without [421] Creutzfeldt-Jakob disease. An increase in Aβ-proteopathy also has been reported in a subset of people who had received cadaveric dura mater transplants [422, 423]. The most parsimonious explanation for these findings is that some batches of therapeutic growth hormone and dura mater were tainted by Aβ seeds that were present in the tissues taken from donors with AD (or incipient AD) [417]. This possibility is reinforced by the demonstrable presence of aggregated Aβ in some pituitary glands [424] and dura mater [425] from AD patients. Furthermore, Aβ was detected in archival samples of cadaveric human growth hormone [426], and stored hormone was shown to stimulate cerebral Aβ deposition when injected intracerebrally into APP-transgenic mice [427]. Interestingly, tauopathy was not apparent in most of these cases (even though some abnormal tau is present in Alzheimeric pituitaries), and no recipients of cadaveric growth hormone or dura mater have yet been found to develop full-blown AD. Whether this will happen as the subjects age further remains to be determined. 10. Aβ plaques in nonhuman species 10.1 Native Aβ plaques Naturally occurring Aβ plaques and/or Aβ-CAA have been identified in aged animals of many species, including such diverse creatures as woodpeckers [428], bears [429-431], dogs [432-435], cats [436], camels [437] wolverines [438], and all species of nonhuman primate examined to date [165, 435, 439]. The mammalian mainstays of experimental biology - rats and mice - do not normally manifest plaques in old age, possibly owing to 3 amino acid differences in the N-terminal segment of Aβ that render the protein less likely to aggregate [440, 441]. Most research on native Aβ plaques in nonhuman species has focused on primates ranging from prosimians to monkeys and apes [439], work that has yielded insights into the pathobiology of the lesions [178, 439, 442-444]. Nonhuman primates express Aβ with the same sequence of amino acids as in humans, and both diffuse and dense-core Aβ plaques can be abundant in aged primates [439] (Figure 22). Some of the plaques include reactive glial cells and dysmorphic neurites [444]. Mass-spectrometry has shown that post-translational modifications of Aβ are similar in humans and squirrel monkeys (Saimiri sciureus), and by ELISA, the amount of Aβ in the nonhuman primate brain sometimes exceeds that in humans with AD [163]. Despite the presence of copious aberrant Aβ, no nonhuman species has yet been found to exhibit the full clinicopathologic phenotype of AD as it occurs in humans [165]. Specifically, a dementia-like condition has not yet been identified, and tauopathy, though often present, is generally mild, even in the presence of profuse Aβ plaques. For unknown reasons, Aβ-CAA, especially capillary Aβ-CAA, is more commonly present in nonhuman primates than in humans [439, 445, 446]. Although congophilic Aβ plaques occur, human-like classical plaques with an Aβ core, space, and outer corona (see Figure 1) are rare, if they can be found at all, in prosimians and monkeys (we cannot yet rule out such lesions in great apes, as relatively few have been examined in advanced old age). Surprisingly, there is little high-affinity binding of the Aβ-amyloid-imaging agent Pittsburgh Compound-B (PiB) to Aβ plaques in nonhuman primates, suggesting biochemical and/or conformational differences in the protein between humans and other primates [447]. It is necessary to determine how differences in lifespan and environmental and genetic risk factors might influence the apparent species-specificity of AD and the Aβ-deposition phenotype. However, current evidence suggests that, despite similarities in the sequence, expression, modification, and deposition of Aβ, nonhuman primates lack the permissive connection between Aβ-proteopathy and tauopathy that is critical to the occurrence of AD in humans [165]. Clarifying the nature of this naturally occurring interruption of the Aβ cascade in nonhuman species could reveal new pathogenic pathways for therapeutic intervention in AD.
Figure 22. Aβ deposition (left) in the superior temporal gyrus and a neuritic plaque (right) in the hippocampal formation of two aged rhesus monkeys (Macaca mulatta; 35 years and ~30 years, respectively). Left: Antibody 82E1 to the N-terminal segment of Aβ, Nissl counterstain; Right: Antibody 06-17 to phosphorylated neurofilaments. Bars = 200μm (left) and 25μm (right). The maximum known lifespan of rhesus monkeys is 44 years (see Stonebarger et al. [2020] [reference 512]).
Figure 23. Aβ plaques in an aged (28 months) Tg2576 APP-transgenic mouse. Diffuse deposits are black, and some dense deposits have a golden core (one in the frontal cortex is magnified at right). Campbell-Gallyas silver stain. Bars = 1mm (left) and 50μm (right). 10.2 Aβ plaques in genetically modified animals Studies of naturally occurring plaques in various species have shed some light on the lesions, but there was no practical model in which plaques could be experimentally investigated until the mid-1990s. Then, transgenic mice were introduced that overexpress human APP with genetic mutations linked to AD [448-450]. With age, these APP-transgenic mice deposit copious Aβ in the brain (Figure 23). They were followed by a wealth of additional models in various mouse (and later rat) strains with diverse genetic alterations, transgene expression levels, and the expression or deletion of interacting molecules [451-453]; see Alzforum for a list of rodent models of AD-like pathology: https://www.alzforum.org/research-models/alzheimers-disease). Not surprisingly, the sundry genetically modified animals exhibit many plaque (and CAA) phenotypes. No genetically modified rodent, including those with multiple modifications, has manifested fully AD-like Aβ plaques. As in nonhuman primates, the core-space-corona type of plaque is not typical of the transgenic rodent models. The plaques do, however, share several key features with those in humans; they exhibit a range of morphologies, many have bona fide amyloid cores, and they are invested by aberrant neurites and glial cells [452]. Tau abnormalities occur, but human-like neurofibrillary tangles have not yet been generated in rodents. Nonvertebrate transgenic animals such as fruit flies (Drosophila melanogaster) [454-456] and roundworms (Caenorhabditis elegans) [457, 458] have been developed to study the pathobiology of Aβ. These models can be useful for analyzing molecular mechanisms and for studying the early-stage efficacy and toxicity of investigational agents, but no nonvertebrate model has yet generated Aβ plaques that remotely resemble those in humans. It is difficult to overstate the impact that the introduction of genetically modified animals has had on the course of research on the mechanisms underlying plaque formation and AD. For example, transgenic rodents were used to establish the prion paradigm as the pre-eminent theory of plaque ontogeny and spread [217, 218, 407] (see Section 9), they are being used to probe the role of glial cells and neuritic dystrophy in plaque pathophysiology (see Section 6), and they are a vital tool in the preclinical testing of new therapeutic and diagnostic strategies [452, 459-463]. For instance, whereas the first evidence that Aβ plaques and Aβ-CAA could be targeted by anti-Aβ antibodies in the living brain came from experiments in nonhuman primates [464], genetically modified mice enabled the development of Aβ-immunization as a strategy for the prevention or treatment of AD [465]. The application of longitudinal, in vivo-imaging studies in murine models has facilitated unique insights into the dynamics of Aβ plaques and their cellular constituents (e.g., [323, 324, 466-469]), as well as the response of plaques and Aβ-CAA to therapeutic intervention [451, 452, 470]. Currently, genetically modified nonhuman primates are being created with the hope that they will more completely recapitulate a human-like AD phenotype [471, 472], but no histopathologic findings have yet been reported. Despite the limitation that genetically modified animals do not yet fully recapitulate AD, they will continue to play an important part in deciphering the pathobiology of Aβ plaques. 11. Conclusions: Aβ plaques as a therapeutic objective Aβ plaques are an obligatory component of the pathobiology of AD, and as such, strategies to reduce or neutralize plaques intersect with general strategies to prevent or treat AD. However, the value of plaques, in and of themselves, as therapeutic targets is uncertain. There is little question that Aβ plaques, especially in their more elaborate states, are deleterious to brain tissue; they disrupt neuronal processes and synapses, they can be a source of harmful Aβ-oligomers, and local glial cells create a toxic inflammatory environment. Therapeutically targeting plaques thus presents both opportunity and obstacles. First, given the long, pre-symptomatic emergence and proliferation of Aβ plaques (and neurofibrillary tangles) in the brain [109, 473], as well as evidence of extensive brain damage by the time dementia sets in, early prevention is likely to be the most effective strategy for subduing AD [474, 475]. The promise of prevention is underscored by the protective effects of the A673T mutation in APP, which diminishes Aβ production throughout life and lowers the risk of AD [48]. Since the most effective preventive protocol should be initiated years, and possibly decades, before the predicted onset of dementia, testing for long-term safety and efficacy will be challenging. Additionally, it is not known when, in the course of life, therapy must begin to effectively prevent or delay AD. Second, it is possible that some, if not most, of the direct toxic influence of the Aβ is mediated by oligomeric Aβ rather than by fibrillar amyloid per se. Evidence that Aβ plaques can serve as a source of oligomers (Section 1.1) argues that some benefit can be achieved by reducing plaque burden and thus the accompanying oligomers. It is encouraging that several of the more promising antibodies currently in clinical trials for AD show activity against oligomeric Aβ [475, 476]. A recent study in mice indicates that the short-term neutralization of oligomeric Aβ seeds early in life diminishes plaque formation as the animals age [477]. However, whether mitigating the production, seeding potential or toxicity of oligomeric Aβ will be beneficial in humans, either as a preventive or as a treatment for discernible dementia, remains to be determined. It is also important to consider the possibility that treatments that block Aβ-amyloid fibril assembly, or that disassemble plaques, might inadvertently increase the presence of toxic oligomers. Third, the inability of anti-Aβ immunotherapies to substantially impede dementia in symptomatic subjects, even when Aβ plaques are reduced in number [336, 476, 478, 479], suggests that the dis-integration of the cerebral connectome caused by plaques and tangles is pronounced and largely irreversible once dementia commences [9, 474]. Furthermore, tauopathy is an essential contributor to dementia that itself progresses by a prion-like mechanism [480, 481] that may be at least partly independent of Aβ-proteopathy [336]. Whether the alternative approach of lowering the inflammatory state associated with plaques will meaningfully improve behavior at this later stage of disease also has not been established. In short, once Aβ-amyloid plaques and tauopathy become widespread, especially in neocortical regions [1], removing the plaques is unlikely to significantly reverse the course of dementia. Even so, there is evidence that a reduction of tauopathy [336], and possibly some cognitive benefit, can be achieved in symptomatic patients by anti-Aβ immunotherapy [476, 482-484]. Indeed, active immunization with AN1792, which targets both Aβ plaques and oligomers, resulted in a long-term decrease in all components of the plaques - aggregated Aβ, aberrant neurites, tauopathy, and focal gliopathy - along with improved indices of ‘neuronal health’ [336]. Thus, notwithstanding the mostly disheartening outcome of therapeutic trials to date, current preclinical and clinical data indicate that the right anti-Aβ treatment, at the right time, has a good chance of delaying or preventing AD. Finally, it is imperative to remain vigilant to the impact of environmental factors and the microbiome [485, 486] on the risk of developing AD. For instance, if specific microbes are convincingly found to increase the risk of plaque formation and AD, early immunization against this infectious agent could be an effective preventive measure. It is important also to consider the possibility that interactions among genetic, microbiomic and/or environmental influences could raise disease risk well above the additive impact of individual risk factors. The search for disease-modifying therapies for AD is a broad and rapidly evolving endeavor that has been extensively reviewed (see, e.g., [9, 33, 476, 479, 487-491]. In addition to small molecules, we have entered a phase in medicine in which biologics such as antibodies [492, 493] and nucleic acid-based agents [494, 495] have unprecedented potential to treat neurodegenerative diseases. For well over a century, Aβ plaques have been recognized as an important correlate of dementia in the aging brain. Revealing the mechanisms by which plaques arise, proliferate, and interact with molecular and cellular elements in the nervous system will continue to yield insights into both the ontogeny and treatment of AD. 12. Methods Tissue samples were collected from human subjects with end-stage AD (Figures 1-6, 8-20) and from aged nonhuman species with cerebral Aβ deposition (Figures 21-23). Postmortem collection of samples by the Emory University Goizueta Alzheimer’s Disease Research Center Brain Bank was approved by the Emory Institutional Review Board. Tissues from mice and monkeys were collected at Emory’s Yerkes National Primate Research Center in accordance with federal and institutional guidelines for the humane care and use of experimental animals. The Yerkes Center is fully accredited by AAALAC International. 12.1 Immunohistochemistry For light-microscopy, tissue blocks were embedded, sectioned at 8-10 μm thickness, and mounted onto glass slides for staining. The following antibodies were used for immunohistochemistry: 4G8, mouse monoclonal antibody from Covance (Princeton, NJ) raised against residues 17-24 of Aβ peptide, with an epitope at residues 18-22 [496]; 6E10, mouse monoclonal antibody from Covance raised against residues 1-16 of the Aβ peptide, with an epitope at residues 3-8 [496]; Rabbit polyclonal antibodies R361 and R398, kindly provided by Dr. Pankaj Mehta (Institute for Basic Research on Developmental Disabilities, Staten Island, NY), were raised against synthetic Aβ32-40 and Aβ33-42, respectively [497]; 82E1, mouse monoclonal antibody raised against residues 1-16 of synthetic Aβ [498], from IBL (Gunma, Japan); CP13, mouse monoclonal antibody kindly provided by Dr. Peter Davies (Feinstein Institutes for Medical Research, Manhasset, NY), was raised against a synthetic peptide representing the region around phosphorylated serine residue 202 on the tau protein [499]; MC1, mouse monoclonal antibody, also from Dr. Davies, was raised against Alz50-immunopurified paired helical filaments and then epitope-mapped to similar conformation-specific regions as Alz50 [500]; anti-GFAP, purified immunoglobulin fraction of rabbit antiserum from Dako (Carpinteria, CA) (catalog No. Z0334), raised against GFAP isolated from cow spinal cord and purified by solid-phase absorption with human and cow serum proteins; anti-Iba1, rabbit polyclonal antibody from Wako (Osaka, Japan), raised against a synthetic peptide corresponding to the C-terminus of ionized calcium-binding adapter molecule 1 (Iba1), a 17-kDa protein that is specifically expressed in macrophages/microglia and is upregulated during the activation of these cells [501, 502]; SMI-31, mouse monoclonal antibody raised against a phosphorylated epitope on the neurofilament heavy subunit (NF-H) [503] from BioLegend (San Diego, CA); and 06-17, mouse monoclonal antibody to a phosphorylated epitope shared by the heavy and medium kDa neurofilament polypeptides (generous gift of Drs. Ludwig and Nancy Sternberger, University of Maryland, Baltimore) [504, 505]. Vectastain Elite kits (Vector Laboratories, Burlingame, CA) were used for ABC-based immunodetection of antigen-antibody complexes according to the manufacturer’s instructions, with diaminobenzidine (DAB) as coloring agent for images in Figures 1, 2, 5, 6, 8-11, 14, 16, 17, 19, 21, & 22. In most cases, a Nissl counterstain was applied after immunostaining, as noted. For dual fluorescence immunostaining (Figure 15), the section was incubated in mouse monoclonal antibody CP13 (diluted in 2% normal goat serum) overnight at 4°C, rinsed, and then incubated for 90 minutes in Cy2-conjugated anti-mouse secondary antibody (green; Jackson Labs, West Grove, PA). The section was rinsed thoroughly, incubated overnight in diluted rabbit polyclonal antibody R398 at 4°C, rinsed, and placed for 90 minutes in Rhodamine-Red-X goat anti-rabbit secondary antibody (Jackson Labs). For dual immunostaining by standard transillumination light microscopy (Figure 3), antibodies were sequentially applied as described above except that the polyclonal anti-Aβ antibodies R398+R361 were colored with DAB (brown), and the anti-tau monoclonal antibody CP13 was colored with VIP (purple; Vector Laboratories). Non-immune mouse IgG or rabbit sera were used in place of the primary antibodies as negative controls. Tissues shown in Figures 1 (left) and 4 were stained with the Naoumenko-Feigin silver stain [506] followed by a periodic acid-Schiff (PAS) counterstain. Figure 23 was stained with the Campbell-Gallyas silver stain [507]. Light-microscopic photomicrographs were taken with a Leica DMLB or DMLS microscope (Wetzlar, Germany) and a SPOT FLEX (Diagnostic Instruments, Sterling Heights, MI) or Moticam 5+ (Motic, Hong Kong) digital camera. 12.2 Electron microscopy For conventional ultrastructural analysis (Figures 12, 13, 18, 20), small blocks of cortex were sub-dissected from larger, autopsy-derived tissue blocks that had been immersion-fixed in 10% neutral buffered formalin. The tissue samples were washed in phosphate buffer (0.1M, pH 7.4) and immersed in osmium tetroxide (1% in phosphate buffer) for 20 minutes. They were then rinsed in phosphate buffer and dehydrated in a graded series of ethanol and propylene oxide. Uranyl acetate (1%) was added to the 70% ethanol (35 minute immersion) to improve contrast in the electron microscope. The sections were then embedded in epoxy resin (Durcupan ACM; Fluka, Ft. Washington, PA) on microscope slides and heated for 48 hours at 60°C. Areas of interest were selected, excised from the slide and glued onto resin blocks. Ultrathin sections were cut with a Leica Ultracut T2 (Nussloch, Germany), collected onto single-slot copper grids, and stained with lead citrate. For immunogold EM (Figure 11, right), sections were preincubated in PBS containing 5% nonfat dry milk and then washed in Tris-buffered saline (TBS)-gelatin buffer (0.02 M Tris, 0.15 M NaCl, 1 μl/ml fish gelatin, pH 7.6) to block nonspecific sites. Sections were then incubated for 48 hours at 4°C with antibody 4G8 diluted in PBS-BSA, rinsed in TBS-gelatin, and incubated for 2 hours at room temperature in gold-conjugated goat anti-mouse Fab’ fragments (dilution 1:100; Nanogold [Nanoprobes Inc., Yaphank, NY]). Gold particles were silver-enhanced with the HQ Silver kit (Nanoprobes). The tissue was then embedded and cut as described above. Thin sections were examined with a Zeiss EM10-C electron microscope (Oberkochen, Germany) and digital images were captured using a Dual View camera (Gatan Inc., Pleasanton, CA). Acknowledgements I gratefully acknowledge enlightening discussions with Mathias Jucker, Dietmar Thal, Marla Gearing, Harry LeVine, Rebecca Rosen, Amaryllis Cintron, Eric Heuer, David Lynn, Yury Chernoff, Rolf Warzok, Silke Vogelgesang, Sanjeev Gumber, Linda Cork, Allan Levey and James Lah. Hailian Xiao, Jean-François Paré, and Jeromy Dooyema provided expert technical assistance. Portions of this work were supported by the MetLife Foundation, CART Foundation, Alexander von Humboldt Stiftung, and by National Institutes of Health (NIH) grants P01 AG026423, P50 AG025688, R21 NS077049, and ORIP/OD P51 OD011132. References 1. Nelson, P.T., et al., Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol, 2012. 71(5): p. 362-81. 2. Glenner, G.G. and C.W. Wong, Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun, 1984. 122(3): p. 1131-5. 3. Glenner, G.G. and C.W. Wong, Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun, 1984. 120(3): p. 885-90. 4. Masters, C.L., et al., Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A, 1985. 82(12): p. 4245-9. 5. Masters, C.L. and K. Beyreuther, Pathways to the discovery of the Abeta amyloid of Alzheimer’s disease. J Alzheimers Dis, 2006. 9(3 Suppl): p. 155-61. 6. Benson, M.D., et al., Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid, 2018. 25(4): p. 215-219. 7. Selkoe, D.J., Biology of β-amyloid precursor protein and the mechanism of Alzheimer disease, in Alzheimer Disease, R.D. Terry, et al., Editors. 1999, Lippincott, Williams, and Wilkins Philadelphia. p. 293-310. 8. Haass, C., et al., Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med, 2012. 2(5): p. a006270. 9. Long, J.M. and D.M. Holtzman, Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell, 2019. 179(2): p. 312-339. 10. Dunys, J., A. Valverde, and F. Checler, Are N- and C-terminally truncated Abeta species key pathological triggers in Alzheimer’s disease? J Biol Chem, 2018. 293(40): p. 15419-15428. 11. Kummer, M.P. and M.T. Heneka, Truncated and modified amyloid-beta species. Alzheimers Res Ther, 2014. 6(3): p. 28. 12. Portelius, E., et al., Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol, 2010. 120(2): p. 185-93. 13. Portelius, E., et al., Brain amyloid-beta fragment signatures in pathological ageing and Alzheimer’s disease by hybrid immunoprecipitation mass spectrometry. Neurodegener Dis, 2015. 15(1): p. 50-7. 14. Saido, T.C., et al., Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett, 1996. 215(3): p. 173-6. 15. Takami, M., et al., gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci, 2009. 29(41): p. 13042-52. 16. Tekirian, T.L., et al., N-terminal heterogeneity of parenchymal and cerebrovascular Abeta deposits. J Neuropathol Exp Neurol, 1998. 57(1): p. 76-94. 17. Moro, M.L., et al., Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol Commun, 2018. 6(1): p. 3. 18. Schaffert, L.N. and W.G. Carter, Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci, 2020. 10(4): p. 232. 19. Thal, D.R., et al., Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer’s disease. Acta Neuropathol, 2015. 129(2): p. 167-82. 20. Ke, P.C., et al., Half a century of amyloids: past, present and future. Chem Soc Rev, 2020. 49(15): p. 5473-5509. 21. Greenberg, S.M., et al., Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol, 2020. 16(1): p. 30-42. 22. Holtzman, D.M., J.C. Morris, and A.M. Goate, Alzheimer’s disease: the challenge of the second century. Sci Transl Med, 2011. 3(77): p. 77sr1. 23. Roher, A.E., et al., beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A, 1993. 90(22): p. 10836-40. 24. Attems, J., F. Lintner, and K.A. Jellinger, Amyloid beta peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol, 2004. 107(4): p. 283-91. 25. Richard, E., et al., Characteristics of dyshoric capillary cerebral amyloid angiopathy. J Neuropathol Exp Neurol, 2010. 69(11): p. 1158-67. 26. Attems, J., et al., Capillary CAA and perivascular Abeta-deposition: two distinct features of Alzheimer’s disease pathology. J Neurol Sci, 2010. 299(1-2): p. 155-62. 27. Catalano, S.M., et al., The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer’s disease. Curr Top Med Chem, 2006. 6(6): p. 597-608. 28. Kuo, Y.M., et al., Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem, 1996. 271(8): p. 4077-81. 29. Cline, E.N., et al., The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis, 2018. 64(s1): p. S567-S610. 30. Ferreira, S.T., et al., Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci, 2015. 9: p. 191. 31. Haass, C. and D.J. Selkoe, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol, 2007. 8(2): p. 101-12. 32. Tomiyama, T. and H. Shimada, APP Osaka Mutation in Familial Alzheimer’s Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance. Int J Mol Sci, 2020. 21(4): p. 1413. 33. Walsh, D.M. and D.J. Selkoe, Amyloid beta-protein and beyond: the path forward in Alzheimer’s disease. Curr Opin Neurobiol, 2020. 61: p. 116-124. 34. Flagmeier, P., et al., Direct measurement of lipid membrane disruption connects kinetics and toxicity of Abeta42 aggregation. Nat Struct Mol Biol, 2020. 27(10): p. 886-891. 35. Chen, X.Q. and W.C. Mobley, Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Abeta and Tau Species. Front Neurosci, 2019. 13: p. 659. 36. Esparza, T.J., et al., Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol, 2013. 73(1): p. 104-19. 37. Shankar, G.M., et al., Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med, 2008. 14(8): p. 837-42. 38. Hyman, B.T., Anatomical changes underlying dementia in Alzheimer’s disease, in Alzheimer: 100 Years and Beyond, M. Jucker, et al., Editors. 2006, Springer-Verlag: Berlin Heidelberg New York. p. 89-94. 39. Koffie, R.M., et al., Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A, 2009. 106(10): p. 4012-7. 40. Katzmarski, N., et al., Abeta oligomers trigger and accelerate Abeta seeding. Brain Pathol, 2020. 30(1): p. 36-45. 41. Langer, F., et al., Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci, 2011. 31(41): p. 14488-95. 42. Benilova, I., et al., Highly infectious prions are not directly neurotoxic. Proc Natl Acad Sci U S A, 2020. 117(38): p. 23815-23822. 43. Benilova, I., E. Karran, and B. De Strooper, The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci, 2012. 15(3): p. 349-57. 44. Hardy, J. and D.J. Selkoe, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 2002. 297(5580): p. 353-6. 45. Walker, L.C., D.G. Lynn, and Y.O. Chernoff, A standard model of Alzheimer’s disease? Prion, 2018. 12(5-6): p. 261-265. 46. Wiseman, F.K., et al., A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci, 2015. 16(9): p. 564-74. 47. Abrahamson, E.E., et al., Neuropathological correlates of amyloid PET imaging in Down syndrome. Dev Neurobiol, 2019. 79(7): p. 750-766. 48. Jonsson, T., et al., A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature, 2012. 488(7409): p. 96-9. 49. Benilova, I., et al., The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-beta (Abeta) aggregation. J Biol Chem, 2014. 289(45): p. 30977-89. 50. Kero, M., et al., Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol Aging, 2013. 34(5): p. 1518 e1-3. 51. Di Fede, G., et al., A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science, 2009. 323(5920): p. 1473-7. 52. Buxbaum, J.N. and R.P. Linke, A molecular history of the amyloidoses. J Mol Biol, 2012. 421(2-3): p. 142-59. 53. Puchtler, H. and F. Sweat, A review of early concepts of amyloid in context with contemporary chemical literature from 1839 to 1859. J Histochem Cytochem, 1966. 14(2): p. 123-34. 54. Sipe, J.D. and A.S. Cohen, Review: history of the amyloid fibril. J Struct Biol, 2000. 130(2-3): p. 88-98. 55. Yakupova, E.I., et al., Congo Red and amyloids: history and relationship. Biosci Rep, 2019. 39(1): p. BSR20181415. 56. Chiti, F. and C.M. Dobson, Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem, 2006. 75: p. 333-66. 57. Fandrich, M., On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell Mol Life Sci, 2007. 64(16): p. 2066-78. 58. Westermark, G.T., et al., Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission. Cold Spring Harb Perspect Med, 2018. 8(1): p. a024323. 59. Eisenberg, D. and M. Jucker, The amyloid state of proteins in human diseases. Cell, 2012. 148(6): p. 1188-203. 60. Hardy, J., Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ‘permissive templating’ as a general mechanism underlying neurodegeneration. Biochem Soc Trans, 2005. 33(Pt 4): p. 578-81. 61. Annamalai, K., et al., Polymorphism of Amyloid Fibrils In Vivo. Angew Chem Int Ed Engl, 2016. 55(15): p. 4822-5. 62. Eisenberg, D.S. and M.R. Sawaya, Structural Studies of Amyloid Proteins at the Molecular Level. Annu Rev Biochem, 2017. 86: p. 69-95. 63. Fandrich, M., J. Meinhardt, and N. Grigorieff, Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion, 2009. 3(2): p. 89-93. 64. Fitzpatrick, A.W.P., et al., Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature, 2017. 547(7662): p. 185-190. 65. Iadanza, M.G., et al., The structure of a beta2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat Commun, 2018. 9(1): p. 4517. 66. Kollmer, M., et al., Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun, 2019. 10(1): p. 4760. 67. Petkova, A.T., et al., Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science, 2005. 307(5707): p. 262-5. 68. Roeder, C., et al., Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat Struct Mol Biol, 2020. 27(7): p. 660-667. 69. Pepys, M.B., Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philos Trans R Soc Lond B Biol Sci, 2001. 356(1406): p. 203-10; discussion 210-1. 70. Pinney, J.H. and P.N. Hawkins, Amyloidosis. Ann Clin Biochem, 2012. 49(Pt 3): p. 229-41. 71. Bridger, J. and N.A. Wright, Amyloidosis, in Oxford Textbook of Pathology, J.O.D. McGee, P.G. Isaacson, and N.A. Wright, Editors. 1992, Oxford University Press: New York. p. 406-412. 72. Chen, D., et al., Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat Commun, 2019. 10(1): p. 2493. 73. Arriagada, P.V., et al., Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology, 1992. 42(3 Pt 1): p. 631-9. 74. Bierer, L.M., et al., Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol, 1995. 52(1): p. 81-8. 75. Crystal, H., et al., Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology, 1988. 38(11): p. 1682-7. 76. Gold, G., et al., Clinical validity of A beta-protein deposition staging in brain aging and Alzheimer disease. J Neuropathol Exp Neurol, 2001. 60(10): p. 946-52. 77. Nagy, Z., et al., Relative roles of plaques and tangles in the dementia of Alzheimer’s disease: correlations using three sets of neuropathological criteria. Dementia, 1995. 6(1): p. 21-31. 78. Wilcock, G.K. and M.M. Esiri, Plaques, tangles and dementia. A quantitative study. J Neurol Sci, 1982. 56(2-3): p. 343-56. 79. Braak, H. and E. Braak, Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 1991. 82(4): p. 239-59. 80. Braak, H. and E. Braak, Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging, 1997. 18(4): p. 351-7. 81. Tomlinson, B.E. and J.A.N. Corsellis, Ageing and the dementias, in Greenfield’s Neuropathology, J.H. Adams, J.A.N. Corsellis, and L.W. Duchen, Editors. 1984, John Wiley and Sons: New York. p. 951-1025. 82. He, Z., et al., Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med, 2018. 24(1): p. 29-38. 83. Stancu, I.C., et al., Models of beta-amyloid induced Tau-pathology: the long and "folded" road to understand the mechanism. Mol Neurodegener, 2014. 9: p. 51. 84. Busche, M.A. and B.T. Hyman, Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci, 2020. 23(10): p. 1183-1193. 85. Cook, H.C., Origins of ... tinctorial methods in histology. J Clin Pathol, 1997. 50(9): p. 716-20. 86. Blocq, P. and G. Marinesco, Sur les lésions et la pathogénie de l’épilepsie dite essentielle. La Semaine Médicale, 1892. p. 445-446. 87. Goedert, M., Oskar Fischer and the study of dementia. Brain, 2009. 132(Pt 4): p. 1102-11. 88. Redlich, E., Ueber miliare Sklerose der Hirnrinde bei seniler Atrophie. Jahrbücher für Psychiatrie und Neurologie, 1898. 17: p. 208-216. 89. Ohry, A. and O. Buda, Teofil Simchowicz (1879-1957): the scientist who coined senile plaques in neuropathology. Rom J Morphol Embryol, 2015. 56(4): p. 1545-8. 90. Crook, R., et al., A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med, 1998. 4(4): p. 452-5. 91. Houlden, H., et al., Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol, 2000. 48(5): p. 806-8. 92. Le, T.V., et al., Cotton wool plaques in non-familial late-onset Alzheimer disease. J Neuropathol Exp Neurol, 2001. 60(11): p. 1051-61. 93. Miki, T., et al., Young adult-onset, very slowly progressive cognitive decline with spastic paraparesis in Alzheimer’s disease with cotton wool plaques due to a novel presenilin1 G417S mutation. Acta Neuropathol Commun, 2019. 7(1): p. 19. 94. Miravalle, L., et al., Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry, 2005. 44(32): p. 10810-21. 95. Alzheimer, A., Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie, 1907. 64: p. 146-148. 96. Alzheimer, A., Über eigenartige Krankheitsfälle des späteren Alters. Zeitschrift für die gesamte Neurologie und Psychiatrie, 1911. 4: p. 356-385. 97. Miyake, K., Beiträge zur Kenntnis der Altersveränderungen der menschilchen Hirnrinde. Arbeiten Obersteinerschen Neurologische Institut Wien, 1906. 13: p. 212-259. 98. Lafora, G.R., Beitrag zur Kenntnis der Alzheimerschen Krankheit oder präsenilen Demenz mit Herdsymptomen. Zeitschrift für die gesamte Neurologie und Psychiatrie, 1911. 6(1): p. 15. 99. Bonfiglio, F., Di speciali reperti in un caso di probabile sifilide cerebrale. Rivista Sperimentale di Feniatria 1908. 34: p. 196-206. 100. Hübner, A.H., Zur Histopathologie der senilen Hirnrinde. Archiv für Psychiatrie und Nervenkrankheiten, 1909. 46(2): p. 598-609. 101. Perusini, G., Über klinisch und histologisch eigenartige psychische Erkrankungen des höheren Lebensalters, in Histologische und Histopathologische Arbeiten über die Grosshirnrinde, F. Nissl and A. Alzheimer, Editors. 1910, Fischer-Verlag: Jena. p. 297-351. 102. Fuller, S.C., A study of the miliary plaques found in brains of the aged. American Journal of Insanity, 1911. 68(2): p. 147-219. 103. Bielschowsky, M., Zur Kenntnis der Alzheimerschen Krankheit (präsenilen Demenz mit Herdsymptomen). Journal für Psychologie und Neurologie 1911. 18: p. 1-20. 104. Barrett, A.M., Degeneration of intracellular neurofibrils with miliary gliosis in psychoses of the senile period. American Journal of Insanity, 1911. 67(3): p. 503-516. 105. Simchowicz, T., Histologische Studien über die senile Demenz, in Histologische und Histopathologische Arbeiten über die Grosshirnrinde, F. Nissl and A. Alzheimer, Editors. 1911, Fischer-Verlag: Jena. p. 267-444. 106. Marinesco, G. and J. Minea, Untersuchungen über die “senilen Plaques”. Monatschrift für Psychiatrie und Neurologie, 1912. 31: p. 79-91. 107. Christen, Y., Alois Alzheimer and the myth of the pioneer, in Alzheimer: 100 Years and Beyond, M. Jucker, et al., Editors. 2006, Springer: Berlin Heidelberg New York. p. 51-55. 108. Braak, H. and E. Braak, Neurofibrillary changes: The hallmark of Alzheimer disease, in Concepts of Alzheimer Disease: Biological, Clinical and Cultural Perspectives, P.J. Whitehouse, K. Mauer, and J. Ballenger, Editors. 2000, Johns Hopkins University Press: Baltimore. p. 53-71. 109. Jack, C.R., Jr., et al., NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement, 2018. 14(4): p. 535-562. 110. Bick, K., L. Amaducci, and G. Pepeu, The Early Story of Alzheimer’s Disease. 1987, Philadelphia: Lippincott Williams and Wilkins. 111. De Strooper, B. and E. Karran, The Cellular Phase of Alzheimer’s Disease. Cell, 2016. 164(4): p. 603-15. 112. Gleichman, A.J. and S.T. Carmichael, Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol Dis, 2020. 142: p. 104957. 113. Heneka, M.T., et al., Neuroinflammation in Alzheimer’s disease. Lancet Neurol, 2015. 14(4): p. 388-405. 114. Newcombe, E.A., et al., Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation, 2018. 15(1): p. 276. 115. Shi, Y. and D.M. Holtzman, Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol, 2018. 18(12): p. 759-772. 116. Critchley, M., Critical Review: The nature and significance of senile plaques. Journal of Neurology and Psychopathology, 1929. 10(38): p. 124-39. 117. Fischer, O., Miliare Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Monatsschrift für Psychiatrie und Neurologie, 1907. 22: p. 361-372. 118. Ferraro, A., The origin and formation of senile plaques. Archives of Neurology and Psychiatry, 1931. 25(5): p. 1042-1062. 119. Soniat, T.L.L., Histogenesis of senile plaques. Archives of Neurology and Psychiatry, 1941. 46(1): p. 101-114. 120. Liss, L., Senile brain changes, histopathology of the ganglion cells. J Neuropathol Exp Neurol, 1960. 19: p. 559-71. 121. Bouman, L., Senile plaques. Brain, 1934. 57(2): p. 128-142. 122. Kidd, M., Alzheimer’s Disease--an Electron Microscopical Study. Brain, 1964. 87: p. 307-20. 123. Terry, R.D., N.K. Gonatas, and M. Weiss, Ultrastructural Studies in Alzheimer’s Presenile Dementia. Am J Pathol, 1964. 44: p. 269-97. 124. Blessed, G., B.E. Tomlinson, and M. Roth, The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry, 1968. 114(512): p. 797-811. 125. Divry, P., Etude histo-chimique des plaques séniles. Journal Belge de Neurologie et de Psychiatrie, 1927. 9: p. 643-657. 126. Dickson, D.W., Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol, 2009. 3(1): p. 1-23. 127. DeArmond, S.J., et al., Neuropathology of prion diseases, in Prion Biology and Diseases, S.B. Prusiner, Editor. 2004, Cold Spring Harbor Laboratory Press: Cold Spring Harbor. p. 777-856. 128. DeArmond, S.J. and S.B. Prusiner, Etiology and pathogenesis of prion diseases. Am J Pathol, 1995. 146(4): p. 785-811. 129. Holton, J.L., et al., Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol, 2001. 158(2): p. 515-26. 130. Vidal, R., et al., A stop-codon mutation in the BRI gene associated with familial British dementia. Nature, 1999. 399(6738): p. 776-81. 131. Holton, J.L., et al., Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J Neuropathol Exp Neurol, 2002. 61(3): p. 254-67. 132. Vidal, R., et al., A decamer duplication in the 3’ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci U S A, 2000. 97(9): p. 4920-5. 133. Serrano-Pozo, A., et al., Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 2011. 1(1): p. a006189. 134. Steiner, H., et al., A pathogenic presenilin-1 deletion causes abberrant Abeta 42 production in the absence of congophilic amyloid plaques. J Biol Chem, 2001. 276(10): p. 7233-9. 135. Iwatsubo, T., et al., Full-length amyloid-beta (1-42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am J Pathol, 1996. 149(6): p. 1823-30. 136. Lalowski, M., et al., The "nonamyloidogenic" p3 fragment (amyloid beta17-42) is a major constituent of Down’s syndrome cerebellar preamyloid. J Biol Chem, 1996. 271(52): p. 33623-31. 137. The American Heritage Dictionary of the English Language. 2011, Houghton Mifflin Harcourt: Boston, MA. 138. Hauw, J.-J. and C. Duyckaerts, Alzheimer’s disease, in Pathology of the Aging Human Nervous System, S. Duckett and J.C. De La Torre, Editors. 2001, Oxford University Press: New York. p. 207-263. 139. Dickson, D.W., The pathogenesis of senile plaques. J Neuropathol Exp Neurol, 1997. 56(4): p. 321-39. 140. Cupidi, C., et al., Neocortical variation of Abeta load in fully expressed, pure Alzheimer’s disease. J Alzheimers Dis, 2010. 19(1): p. 57-68. 141. Rogers, J. and J.H. Morrison, Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer’s disease. J Neurosci, 1985. 5(10): p. 2801-8. 142. Bero, A.W., et al., Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci, 2011. 14(6): p. 750-6. 143. Wisniewski, H.M., et al., Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol, 1989. 78(4): p. 337-47. 144. Thal, D.R., et al., The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ, 2006. 2006(6): p. re1. 145. Thal, D.R., et al., Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 2002. 58(12): p. 1791-800. 146. Braak, H., et al., Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol, 2011. 70(11): p. 960-9. 147. Grothe, M.J., et al., In vivo staging of regional amyloid deposition. Neurology, 2017. 89(20): p. 2031-2038. 148. Ogomori, K., et al., Beta-protein amyloid is widely distributed in the central nervous system of patients with Alzheimer’s disease. Am J Pathol, 1989. 134(2): p. 243-51. 149. Klunk, W.E., et al., Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol, 2004. 55(3): p. 306-19. 150. Shoghi-Jadid, K., et al., Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry, 2002. 10(1): p. 24-35. 151. Barrio, J.R., et al., In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci U S A, 2015. 112(16): p. E2039-47. 152. Kepe, V., et al., PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis, 2013. 36(1): p. 145-53. 153. Shin, J., et al., The merits of FDDNP-PET imaging in Alzheimer’s disease. J Alzheimers Dis, 2011. 26 Suppl 3: p. 135-45. 154. Mathis, C.A., et al., Small-molecule PET Tracers for Imaging Proteinopathies. Semin Nucl Med, 2017. 47(5): p. 553-575. 155. Clark, C.M., et al., Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA, 2011. 305(3): p. 275-83. 156. Wong, D.F., et al., In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J Nucl Med, 2010. 51(6): p. 913-20. 157. Sabri, O., et al., Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement, 2015. 11(8): p. 964-74. 158. Vandenberghe, R., et al., 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol, 2010. 68(3): p. 319-29. 159. Ikonomovic, M.D., et al., Post-mortem histopathology underlying beta-amyloid PET imaging following flutemetamol F 18 injection. Acta Neuropathol Commun, 2016. 4(1): p. 130. 160. Salloway, S., et al., Performance of [(18)F]flutemetamol amyloid imaging against the neuritic plaque component of CERAD and the current (2012) NIA-AA recommendations for the neuropathologic diagnosis of Alzheimer’s disease. Alzheimers Dement (Amst), 2017. 9: p. 25-34. 161. Thal, D.R., et al., Different aspects of Alzheimer’s disease-related amyloid beta-peptide pathology and their relationship to amyloid positron emission tomography imaging and dementia. Acta Neuropathol Commun, 2019. 7(1): p. 178. 162. Diner, I., et al., Generation of Clickable Pittsburgh Compound B for the Detection and Capture of beta-Amyloid in Alzheimer’s Disease Brain. Bioconjug Chem, 2017. 28(10): p. 2627-2637. 163. Rosen, R.F., et al., Comparative pathobiology of beta-amyloid and the unique susceptibility of humans to Alzheimer’s disease. Neurobiol Aging, 2016. 44: p. 185-196. 164. Rosen, R.F., et al., Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol, 2010. 119(2): p. 221-33. 165. Walker, L.C. and M. Jucker, The exceptional vulnerability of humans to Alzheimer’s disease. Trends Mol Med, 2017. 23(6): p. 534-545. 166. Ikeda, S., D. Allsop, and G.G. Glenner, Morphology and distribution of plaque and related deposits in the brains of Alzheimer’s disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab Invest, 1989. 60(1): p. 113-22. 167. Ikeda, S., et al., Evidence of amyloid beta-protein immunoreactive early plaque lesions in Down’s syndrome brains. Lab Invest, 1989. 61(1): p. 133-7. 168. Tagliavini, F., et al., Preamyloid deposits in the cerebral cortex of patients with Alzheimer’s disease and nondemented individuals. Neurosci Lett, 1988. 93(2-3): p. 191-6. 169. Tagliavini, F., et al., Cerebral extracellular preamyloid deposits in Alzheimer’s disease, Down syndrome and nondemented elderly individuals. Prog Clin Biol Res, 1989. 317: p. 1001-5. 170. Yamaguchi, H., et al., Diffuse type of senile plaques in the brains of Alzheimer-type dementia. Acta Neuropathol, 1988. 77(2): p. 113-9. 171. Yamaguchi, H., et al., A variety of cerebral amyloid deposits in the brains of the Alzheimer-type dementia demonstrated by beta protein immunostaining. Acta Neuropathol, 1988. 76(6): p. 541-9. 172. Yamaguchi, H., et al., Alzheimer type dementia: diffuse type of senile plaques demonstrated by beta protein immunostaining. Prog Clin Biol Res, 1989. 317: p. 467-74. 173. Armstrong, R.A., Beta-amyloid plaques: stages in life history or independent origin? Dement Geriatr Cogn Disord, 1998. 9(4): p. 227-38. 174. Dickson, T.C. and J.C. Vickers, The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience, 2001. 105(1): p. 99-107. 175. Duyckaerts, C., B. Delatour, and M.C. Potier, Classification and basic pathology of Alzheimer disease. Acta Neuropathol, 2009. 118(1): p. 5-36. 176. Thal, D.R., et al., Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol, 2000. 59(8): p. 733-48. 177. Masters, C.L. and K. Beyreuther, Henryk M. Wisniewski and the amyloid theory of Alzheimer’s disease. J Alzheimers Dis, 2001. 3(1): p. 83-86. 178. Wisniewski, H.M. and R.D. Terry, Reexamination of the pathogenesis of the senile plaque, in Progress in Neuropathology, H.M. Zimmerman, Editor. 1973, Grune & Stratton: New York. p. 1-26. 179. Boon, B.D.C., et al., The coarse-grained plaque: a divergent Abeta plaque-type in early-onset Alzheimer’s disease. Acta Neuropathol, 2020. doi: 10.1007/s00401-020-02198-8. Online ahead of print. PMID: 32926214. 180. Thal, D.R., et al., Fleecy amyloid deposits in the internal layers of the human entorhinal cortex are comprised of N-terminal truncated fragments of Abeta. J Neuropathol Exp Neurol, 1999. 58(2): p. 210-6. 181. Davies, C.A. and D.M. Mann, Is the "preamyloid" of diffuse plaques in Alzheimer’s disease really nonfibrillar? Am J Pathol, 1993. 143(6): p. 1594-605. 182. Yamaguchi, H., et al., Electron micrograph of diffuse plaques. Initial stage of senile plaque formation in the Alzheimer brain. Am J Pathol, 1989. 135(4): p. 593-7. 183. Yamaguchi, H., et al., Ultrastructure of diffuse plaques in senile dementia of the Alzheimer type: comparison with primitive plaques. Acta Neuropathol, 1991. 82(1): p. 13-20. 184. Wisniewski, H.M., et al., Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease. J Neuropathol Exp Neurol, 1998. 57(7): p. 674-83. 185. Fischer, O., Die presbyophrene Demenz, deren anatomische Grundlage und klinische Abgrenzung. Zeitschrift für die gesamte Neurologie und Psychiatrie, 1910. 3: p. 371-471. 186. Thal, D.R., et al., Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol, 2000. 100(6): p. 608-17. 187. Allsop, D., et al., Neurofibrillary tangles in some cases of dementia pugilistica share antigens with amyloid beta-protein of Alzheimer’s disease. Am J Pathol, 1990. 136(2): p. 255-60. 188. Bondareff, W., et al., Molecular analysis of neurofibrillary degeneration in Alzheimer’s disease. An immunohistochemical study. Am J Pathol, 1990. 137(3): p. 711-23. 189. Hyman, B.T., et al., A4 amyloid protein immunoreactivity is present in Alzheimer’s disease neurofibrillary tangles. Neurosci Lett, 1989. 101(3): p. 352-5. 190. Sherriff, F.E., L.R. Bridges, and D.S. De Souza, Non-Alzheimer neurofibrillary tangles show beta-amyloid-like immunoreactivity. Neuroreport, 1994. 5(15): p. 1897-900. 191. Tabaton, M., et al., Ultrastructural localization of beta-amyloid, tau, and ubiquitin epitopes in extracellular neurofibrillary tangles. Proc Natl Acad Sci U S A, 1991. 88(6): p. 2098-102. 192. Yamaguchi, H., et al., Secondary deposition of beta amyloid within extracellular neurofibrillary tangles in Alzheimer-type dementia. Am J Pathol, 1991. 138(3): p. 699-705. 193. Zemlan, F.P., et al., Alzheimer’s paired helical filaments: amyloid precursor protein epitope mapping. Brain Res Bull, 1994. 33(4): p. 387-92. 194. Walker, L.C., et al., Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol, 2000. 100(1): p. 36-42. 195. Mott, R.T. and C.M. Hulette, Neuropathology of Alzheimer’s disease. Neuroimaging Clin N Am, 2005. 15(4): p. 755-65, ix. 196. Edgar, J.R., et al., ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J Cell Sci, 2015. 128(14): p. 2520-8. 197. Langui, D., et al., Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol, 2004. 165(5): p. 1465-77. 198. Rajendran, L., et al., Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A, 2006. 103(30): p. 11172-7. 199. Takahashi, R.H., et al., Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol, 2002. 161(5): p. 1869-79. 200. Willen, K., et al., Abeta accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener, 2017. 12(1): p. 61. 201. Becot, A., C. Volgers, and G. van Niel, Transmissible Endosomal Intoxication: A Balance between Exosomes and Lysosomes at the Basis of Intercellular Amyloid Propagation. Biomedicines, 2020. 8(8). 202. Han, S., et al., Amyloid plaque structure and cell surface interactions of beta-amyloid fibrils revealed by electron tomography. Sci Rep, 2017. 7: p. 43577. 203. Schmidt, M.L., et al., Chemical and immunological heterogeneity of fibrillar amyloid in plaques of Alzheimer’s disease and Down’s syndrome brains revealed by confocal microscopy. Am J Pathol, 1995. 147(2): p. 503-15. 204. Nelson, P.T., H. Braak, and W.R. Markesbery, Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol, 2009. 68(1): p. 1-14. 205. Prusiner, S.B., An introduction to Prion Diseases, in Prion Diseases, S.B. Prusiner, Editor. 2017, Cold Spring Harbor Laboratory Press: Cold Spring Harbor p. 1-29. 206. Will, R.G. and J.W. Ironside, Sporadic and Infectious Human Prion Diseases. Cold Spring Harb Perspect Med, 2017. 7(1): p. a024364. 207. Fiala, J.C., Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol, 2007. 114(6): p. 551-71. 208. Azizeh, B.Y., et al., Molecular dating of senile plaques in the brains of individuals with Down syndrome and in aged dogs. Exp Neurol, 2000. 163(1): p. 111-22. 209. Fonseca, M.I., et al., The presence of isoaspartic acid in beta-amyloid plaques indicates plaque age. Exp Neurol, 1999. 157(2): p. 277-88. 210. Lee, H.G., et al., Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann N Y Acad Sci, 2004. 1019: p. 1-4. 211. Makin, S., The amyloid hypothesis on trial. Nature, 2018. 559(7715): p. S4-S7. 212. Walker, L.C., Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases. Annu Rev Genet, 2016. 50: p. 329-346. 213. Lau, H.H.C., M. Ingelsson, and J.C. Watts, The existence of Abeta strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol, 2020. doi: 10.1007/s00401-020-02201-2. Online ahead of print. PMID: 32743745. 214. Bartz, J.C., Prion Strain Diversity. Cold Spring Harb Perspect Med, 2016. 6(12): p. a024349. 215. Ghaemmaghami, S., Biology and Genetics of PrP Prion Strains. Cold Spring Harb Perspect Med, 2017. 7(8): p. a026922. 216. Prusiner, S.B., Prion Diseases. 2017, Cold Spring Harbor: Cold Spring Harbor Laboratory Press. 217. Jucker, M. and L.C. Walker, Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature, 2013. 501(7465): p. 45-51. 218. Walker, L.C. and M. Jucker, Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci, 2015. 38: p. 87-103. 219. Tanaka, M., et al., The physical basis of how prion conformations determine strain phenotypes. Nature, 2006. 442(7102): p. 585-9. 220. Pedersen, J.S. and D.E. Otzen, Amyloid-a state in many guises: survival of the fittest fibril fold. Protein Sci, 2008. 17(1): p. 2-10. 221. Yagi, H., et al., Visualization and classification of amyloid beta supramolecular assemblies. Biochemistry, 2007. 46(51): p. 15009-17. 222. Mehta, A.K., et al., Context dependence of protein misfolding and structural strains in neurodegenerative diseases. Biopolymers, 2013. 100(6): p. 722-30. 223. Li, J., et al., Darwinian evolution of prions in cell culture. Science, 2010. 327(5967): p. 869-72. 224. Collinge, J. and A.R. Clarke, A general model of prion strains and their pathogenicity. Science, 2007. 318(5852): p. 930-6. 225. Xu, G., et al., Diversity in Abeta deposit morphology and secondary proteome insolubility across models of Alzheimer-type amyloidosis. Acta Neuropathol Commun, 2020. 8(1): p. 43. 226. Cohen, M., B. Appleby, and J.G. Safar, Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion, 2016. 10(1): p. 9-17. 227. Condello, C., et al., Structural heterogeneity and intersubject variability of Abeta in familial and sporadic Alzheimer’s disease. Proc Natl Acad Sci U S A, 2018. 115(4): p. E782-E791. 228. Lu, J.X., et al., Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell, 2013. 154(6): p. 1257-68. 229. Qiang, W., et al., Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature, 2017. 541(7636): p. 217-221. 230. Rasmussen, J., et al., Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc Natl Acad Sci U S A, 2017. 114(49): p. 13018-13023. 231. Watts, J.C., et al., Serial propagation of distinct strains of Abeta prions from Alzheimer’s disease patients. Proc Natl Acad Sci U S A, 2014. 111(28): p. 10323-8. 232. Fitzpatrick, A.W. and H.R. Saibil, Cryo-EM of amyloid fibrils and cellular aggregates. Curr Opin Struct Biol, 2019. 58: p. 34-42. 233. Paravastu, A.K., et al., Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A, 2008. 105(47): p. 18349-54. 234. Sachse, C., M. Fandrich, and N. Grigorieff, Paired beta-sheet structure of an Abeta(1-40) amyloid fibril revealed by electron microscopy. Proc Natl Acad Sci U S A, 2008. 105(21): p. 7462-6. 235. Wang, H., et al., Polymorphic Abeta42 fibrils adopt similar secondary structure but differ in cross-strand side chain stacking interactions within the same beta-sheet. Sci Rep, 2020. 10(1): p. 5720. 236. Revesz, T., et al., Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol, 2003. 62(9): p. 885-98. 237. Biffi, A. and S.M. Greenberg, Cerebral amyloid angiopathy: a systematic review. J Clin Neurol, 2011. 7(1): p. 1-9. 238. Haan, J., et al., Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Arch Neurol, 1990. 47(9): p. 965-7. 239. Maat-Schieman, M.L., et al., Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II--A review of histopathological aspects. Brain Pathol, 1996. 6(2): p. 115-20. 240. Revesz, T., et al., Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol, 2009. 118(1): p. 115-30. 241. Attems, J., Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol, 2005. 110(4): p. 345-59. 242. Attems, J. and K.A. Jellinger, The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med, 2014. 12: p. 206. 243. Kapasi, A. and J.A. Schneider, Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim Biophys Acta, 2016. 1862(5): p. 878-86. 244. Vinters, H.V., Emerging concepts in Alzheimer’s disease. Annu Rev Pathol, 2015. 10: p. 291-319. 245. Auriel, E. and S.M. Greenberg, The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr Atheroscler Rep, 2012. 14(4): p. 343-50. 246. Olichney, J.M., et al., Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol, 1995. 52(7): p. 702-8. 247. Kamara, D.M., et al., Cerebral Amyloid Angiopathy: Similarity in African-Americans and Caucasians with Alzheimer’s Disease. J Alzheimers Dis, 2018. 62(4): p. 1815-1826. 248. Bornebroek, M., et al., Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I--A review of clinical, radiologic and genetic aspects. Brain Pathol, 1996. 6(2): p. 111-4. 249. Kamp, J.A., et al., Amyloid beta in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Rev Neurosci, 2014. 25(5): p. 641-51. 250. Timmers, W.F., et al., Parenchymal preamyloid and amyloid deposits in the brains of patients with hereditary cerebral hemorrhage with amyloidosis--Dutch type. Neurosci Lett, 1990. 118(2): p. 223-6. 251. van Duinen, S.G., et al., Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc Natl Acad Sci U S A, 1987. 84(16): p. 5991-4. 252. Natte, R., et al., Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol, 2001. 50(6): p. 765-72. 253. Wattendorff, A.R., et al., Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J Neurol Neurosurg Psychiatry, 1995. 58(6): p. 699-705. 254. Case, N.F., et al., Cerebral Amyloid Angiopathy Is Associated With Executive Dysfunction and Mild Cognitive Impairment. Stroke, 2016. 47(8): p. 2010-6. 255. Pfeifer, L.A., et al., Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology, 2002. 58(11): p. 1629-34. 256. Schrag, M. and H. Kirshner, Neuropsychological Effects of Cerebral Amyloid Angiopathy. Curr Neurol Neurosci Rep, 2016. 16(8): p. 76. 257. Thal, D.R., et al., Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol, 2003. 62(12): p. 1287-301. 258. Xiong, L., et al., Dementia incidence and predictors in cerebral amyloid angiopathy patients without intracerebral hemorrhage. J Cereb Blood Flow Metab, 2018. 38(2): p. 241-249. 259. Smith, E.E., Cerebral amyloid angiopathy as a cause of neurodegeneration. J Neurochem, 2018. 144(5): p. 651-658. 260. Cisternas, P., X. Taylor, and C.A. Lasagna-Reeves, The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. Int J Mol Sci, 2019. 20(24): p. 6319. 261. Grabowski, T.J., et al., Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol, 2001. 49(6): p. 697-705. 262. Oshima, K., et al., Relative paucity of tau accumulation in the small areas with abundant Abeta42-positive capillary amyloid angiopathy within a given cortical region in the brain of patients with Alzheimer pathology. Acta Neuropathol, 2006. 111(6): p. 510-8. 263. Yamaguchi, H. and M.L.C. Maat-Schieman, Immunohistochemical analysis of amyloid beta-protein isoforms in CAA, in Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders, M.M. Verbeek, R.M.W. de Waal, and H.V. Vinters, Editors. 2000, Kluwer Academic Publishers: Dordrecht. p. 179-188. 264. Roher, A.E., et al., Chemical analysis of Amyloid beta protein in CAA, in Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders, M.M. Verbeek, R.M.W. de Waal, and H.V. Vinters, Editors. 2000, Kluwer Academic Publishers: Dordrecht. p. 157-177. 265. Kawai, M., et al., The relationship of amyloid plaques to cerebral capillaries in Alzheimer’s disease. Am J Pathol, 1990. 137(6): p. 1435-46. 266. Vinters, H.V., Cerebral amyloid angiopathy. A critical review. Stroke, 1987. 18(2): p. 311-24. 267. de Waal, R.M.W. and M.M. Verbeek, Abeta-associated proteins in cerebral amyloid angiopathy, in Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders, M.M. Verbeek, R.M.W. de Waal, and H.V. Vinters, Editors. 2000, Kluwer Academic Publishers: Dordrecht. p. 207-221. 268. Boche, D., et al., Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain, 2008. 131(Pt 12): p. 3299-310. 269. Yamada, M., Cerebral amyloid angiopathy: emerging concepts. J Stroke, 2015. 17(1): p. 17-30. 270. Probst, A., et al., Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res, 1983. 268(2): p. 249-54. 271. Friedrich, R.P., et al., Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A, 2010. 107(5): p. 1942-7. 272. Takahashi, R.H., T. Nagao, and G.K. Gouras, Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol Int, 2017. 67(4): p. 185-193. 273. Sadleir, K.R., et al., Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol, 2016. 132(2): p. 235-256. 274. Kuchibhotla, K.V., et al., Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron, 2008. 59(2): p. 214-25. 275. Sanchez-Varo, R., et al., Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol, 2012. 123(1): p. 53-70. 276. Wirths, O., et al., Lewy body variant of Alzheimer’s disease: alpha-synuclein in dystrophic neurites of A beta plaques. Neuroreport, 2000. 11(17): p. 3737-41. 277. Dickson, T.C., et al., Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp Neurol, 1999. 156(1): p. 100-10. 278. Garcia-Marin, V., P. Garcia-Lopez, and M. Freire, Cajal’s contributions to the study of Alzheimer’s disease. J Alzheimers Dis, 2007. 12(2): p. 161-74. 279. Zhan, S.S., et al., Distribution of neuronal growth-promoting factors and cytoskeletal proteins in altered neurites in Alzheimer’s disease and non-demented elderly. Acta Neuropathol, 1995. 89(4): p. 356-62. 280. Phinney, A.L., et al., Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci, 1999. 19(19): p. 8552-9. 281. Chan-Palay, V., et al., Distribution of altered hippocampal neurons and axons immunoreactive with antisera against neuropeptide Y in Alzheimer’s-type dementia. J Comp Neurol, 1986. 248(3): p. 376-94. 282. Kitt, C.A., et al., Evidence for cholinergic neurites in senile plaques. Science, 1984. 226(4681): p. 1443-5. 283. Kitt, C.A., et al., Catecholaminergic neurites in senile plaques in prefrontal cortex of aged nonhuman primates. Neuroscience, 1985. 16(3): p. 691-9. 284. Kitt, C.A., et al., Serotoninergic neurites in senile plaques in cingulate cortex of aged nonhuman primate. Synapse, 1989. 3(1): p. 12-8. 285. Morrison, J.H., et al., Somatostatin immunoreactivity in neuritic plaques of Alzheimer’s patients. Nature, 1985. 314(6006): p. 90-2. 286. Struble, R.G., et al., Neuropeptidergic systems in plaques of Alzheimer’s disease. J Neuropathol Exp Neurol, 1987. 46(5): p. 567-84. 287. Walker, L.C., et al., Glutamic acid decarboxylase-like immunoreactive neurites in senile plaques. Neurosci Lett, 1985. 59(2): p. 165-9. 288. Armstrong, D.M., et al., Substance P and somatostatin coexist within neuritic plaques: implications for the pathogenesis of Alzheimer’s disease. Neuroscience, 1989. 31(3): p. 663-71. 289. Walker, L.C., et al., Multiple transmitter systems contribute neurites to individual senile plaques. J Neuropathol Exp Neurol, 1988. 47(2): p. 138-44. 290. Price, D.L., et al., Basal forebrain cholinergic systems in Alzheimer’s disease and related dementias. Neuroscience Commentaries, 1982. 1: p. 84-92. 291. Benzing, W.C., E.J. Mufson, and D.M. Armstrong, Alzheimer’s disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res, 1993. 606(1): p. 10-8. 292. DeKosky, S.T. and S.W. Scheff, Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol, 1990. 27(5): p. 457-64. 293. DeKosky, S.T., S.W. Scheff, and S.D. Styren, Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration, 1996. 5(4): p. 417-21. 294. Masliah, E., et al., Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett, 1994. 174(1): p. 67-72. 295. Terry, R.D., et al., Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol, 1991. 30(4): p. 572-80. 296. Koffie, R.M., B.T. Hyman, and T.L. Spires-Jones, Alzheimer’s disease: synapses gone cold. Mol Neurodegener, 2011. 6(1): p. 63. 297. Spires-Jones, T.L. and B.T. Hyman, The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron, 2014. 82(4): p. 756-71. 298. Sims, R., M. Hill, and J. Williams, The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci, 2020. 23(3): p. 311-322. 299. Bohlen, C.J., et al., Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu Rev Genet, 2019. 53: p. 263-288. 300. Hansen, D.V., J.E. Hanson, and M. Sheng, Microglia in Alzheimer’s disease. J Cell Biol, 2018. 217(2): p. 459-472. 301. Neuner, S.M., J. Tcw, and A.M. Goate, Genetic architecture of Alzheimer’s disease. Neurobiol Dis, 2020. 143: p. 104976. 302. Farfara, D., V. Lifshitz, and D. Frenkel, Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease. J Cell Mol Med, 2008. 12(3): p. 762-80. 303. Liddelow, S.A., et al., Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 2017. 541(7638): p. 481-487. 304. Bouvier, D.S. and K.K. Murai, Synergistic actions of microglia and astrocytes in the progression of Alzheimer’s disease. J Alzheimers Dis, 2015. 45(4): p. 1001-14. 305. Nichols, M.R., et al., Inflammatory mechanisms in neurodegeneration. J Neurochem, 2019. 149(5): p. 562-581. 306. Schwabe, T., K. Srinivasan, and H. Rhinn, Shifting paradigms: The central role of microglia in Alzheimer’s disease. Neurobiol Dis, 2020. 143: p. 104962. 307. Serrano-Pozo, A., et al., Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol, 2013. 72(6): p. 462-71. 308. Town, T., V. Nikolic, and J. Tan, The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation, 2005. 2: p. 24. 309. Wyss-Coray, T. and J. Rogers, Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med, 2012. 2(1): p. a006346. 310. Johnson, E.C.B., et al., Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med, 2020. 26(5): p. 769-780. 311. Nagele, R.G., et al., Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging, 2004. 25(5): p. 663-74. 312. Perez-Nievas, B.G. and A. Serrano-Pozo, Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front Aging Neurosci, 2018. 10: p. 114. 313. Bouvier, D.S., et al., High Resolution Dissection of Reactive Glial Nets in Alzheimer’s Disease. Sci Rep, 2016. 6: p. 24544. 314. Smith, A.J., T. Duan, and A.S. Verkman, Aquaporin-4 reduces neuropathology in a mouse model of Alzheimer’s disease by remodeling peri-plaque astrocyte structure. Acta Neuropathol Commun, 2019. 7(1): p. 74. 315. Heppner, F.L., R.M. Ransohoff, and B. Becher, Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci, 2015. 16(6): p. 358-72. 316. Garwood, C.J., et al., Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: a supporting player with a central role. Neuropathol Appl Neurobiol, 2017. 43(4): p. 281-298. 317. Simpson, J.E., et al., Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging, 2010. 31(4): p. 578-90. 318. Mandrekar-Colucci, S. and G.E. Landreth, Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets, 2010. 9(2): p. 156-67. 319. Galatro, T.F., et al., Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci, 2017. 20(8): p. 1162-1171. 320. Zhou, Y., et al., Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med, 2020. 26(1): p. 131-142. 321. Baik, S.H., et al., Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia, 2016. 64(12): p. 2274-2290. 322. Bolmont, T., et al., Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci, 2008. 28(16): p. 4283-92. 323. Fuger, P., et al., Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci, 2017. 20(10): p. 1371-1376. 324. Hefendehl, J.K., et al., Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J Neurosci, 2011. 31(2): p. 624-9. 325. Ahmad, M.H., M. Fatima, and A.C. Mondal, Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci, 2019. 59: p. 6-11. 326. Frost, G.R., L.A. Jonas, and Y.M. Li, Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front Aging Neurosci, 2019. 11: p. 337. 327. Itagaki, S., et al., Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol, 1989. 24(3): p. 173-82. 328. Mrak, R.E., Neuropathology and the neuroinflammation idea. J Alzheimers Dis, 2009. 18(3): p. 473-81. 329. Sheng, J.G., R.E. Mrak, and W.S. Griffin, Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol, 1997. 94(1): p. 1-5. 330. Vonsattel, J.P. and E.T. Hedley-Whyte, Dementia, in Pathology of the Aging Human Nervous System, S. Duckett and J.C. De La Torre, Editors. 2001, Oxford University Press: New York. p. 156-206. 331. Gratuze, M., C.E.G. Leyns, and D.M. Holtzman, New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener, 2018. 13(1): p. 66. 332. Yeh, F.L., D.V. Hansen, and M. Sheng, TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol Med, 2017. 23(6): p. 512-533. 333. El Khoury, J., et al., Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med, 2007. 13(4): p. 432-8. 334. Wyss-Coray, T., et al., Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A, 2002. 99(16): p. 10837-42. 335. Wilcock, D.M., et al., Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis, 2004. 15(1): p. 11-20. 336. Nicoll, J.A.R., et al., Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain, 2019. 142(7): p. 2113-2126. 337. Keren-Shaul, H., et al., A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 2017. 169(7): p. 1276-1290 e17. 338. Parhizkar, S., et al., Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci, 2019. 22(2): p. 191-204. 339. Leyns, C.E.G., et al., TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci, 2019. 22(8): p. 1217-1222. 340. Frackowiak, J., et al., Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol, 1992. 84(3): p. 225-33. 341. Wisniewski, H.M., et al., Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl, 1990. 7: p. 287-97. 342. Stalder, M., et al., Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol, 1999. 154(6): p. 1673-84. 343. Spangenberg, E., et al., Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun, 2019. 10(1): p. 3758. 344. Hashemiaghdam, A. and M. Mroczek, Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J Neuroimmunol, 2020. 341: p. 577185. 345. Paasila, P.J., et al., Clustering of activated microglia occurs before the formation of dystrophic neurites in the evolution of Aβ plaques in Alzheimer’s disease. Free Neuropathology, 2020. 1(20): p. 1-18. 346. El Hajj, H., et al., Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J Neuroinflammation, 2019. 16(1): p. 87. 347. Streit, W.J., H. Khoshbouei, and I. Bechmann, Dystrophic microglia in late-onset Alzheimer’s disease. Glia, 2020. 68(4): p. 845-854. 348. Smith, A.M. and M. Dragunow, The human side of microglia. Trends Neurosci, 2014. 37(3): p. 125-35. 349. Geirsdottir, L., et al., Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell, 2019. 179(7): p. 1609-1622 e16. 350. Phatnani, H. and T. Maniatis, Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol, 2015. 7(6): p. a020628. 351. Thal, D.R., The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp Neurol, 2012. 236(1): p. 1-5. 352. Frost, G.R. and Y.M. Li, The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol, 2017. 7(12): p. 170228. 353. Funato, H., et al., Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol, 1998. 152(4): p. 983-92. 354. Nagele, R.G., et al., Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res, 2003. 971(2): p. 197-209. 355. Habib, N., et al., Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci, 2020. 23(6): p. 701-706. 356. Matias, I., J. Morgado, and F.C.A. Gomes, Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front Aging Neurosci, 2019. 11: p. 59. 357. Arranz, A.M. and B. De Strooper, The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol, 2019. 18(4): p. 406-414. 358. Holtzman, D. and J. Ulrich, Senescent glia spell trouble in Alzheimer’s disease. Nat Neurosci, 2019. 22(5): p. 683-684. 359. Zhang, P., et al., Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci, 2019. 22(5): p. 719-728. 360. Atwood, C.S., et al., Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides, 2002. 23(7): p. 1343-50. 361. Friede, R.L., Enzyme histochemical studies of senile plaques. Journal of Neuropathology and Experimental Neurology, 1965. 24(3): p. 477-491. 362. McGeer, P.L., et al., Pathological proteins in senile plaques. Tohoku J Exp Med, 1994. 174(3): p. 269-77. 363. Rebeck, G.W., et al., Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the alpha 2-macroglobulin receptor/low-density-lipoprotein receptor-related protein. Ann Neurol, 1995. 37(2): p. 211-7. 364. Stewart, K.L. and S.E. Radford, Amyloid plaques beyond Abeta: a survey of the diverse modulators of amyloid aggregation. Biophys Rev, 2017. 9(4): p. 405-419. 365. Pepys, M.B., et al., Amyloid P component. A critical review. Amyloid, 1997. 4(4): p. 274-295. 366. Coria, F., et al., Isolation and characterization of amyloid P component from Alzheimer’s disease and other types of cerebral amyloidosis. Lab Invest, 1988. 58(4): p. 454-8. 367. Kalaria, R.N. and I. Grahovac, Serum amyloid P immunoreactivity in hippocampal tangles, plaques and vessels: implications for leakage across the blood-brain barrier in Alzheimer’s disease. Brain Res, 1990. 516(2): p. 349-53. 368. Snow, A.D., et al., The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol, 1988. 133(3): p. 456-63. 369. Snow, A.D., et al., Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol, 1990. 137(5): p. 1253-70. 370. Eikelenboom, P., et al., Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol, 1989. 56(4): p. 259-62. 371. Eikelenboom, P. and F.C. Stam, Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol, 1982. 57(2-3): p. 239-42. 372. Ishii, T. and S. Haga, Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol, 1984. 63(4): p. 296-300. 373. Rogers, J., et al., Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A, 1992. 89(21): p. 10016-20. 374. Namba, Y., et al., Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res, 1991. 541(1): p. 163-6. 375. Nishiyama, E., et al., Distribution of apolipoprotein E in senile plaques in brains with Alzheimer’s disease: investigation with the confocal laser scan microscope. Brain Res, 1997. 750(1-2): p. 20-4. 376. Abraham, C.R., D.J. Selkoe, and H. Potter, Immunochemical identification of the serine protease inhibitor alpha 1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell, 1988. 52(4): p. 487-501. 377. Sasaki, N., et al., Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol, 1998. 153(4): p. 1149-55. 378. Smith, M.A., et al., Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A, 1994. 91(12): p. 5710-4. 379. Roy, E.R., et al., Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest, 2020. 130(4): p. 1912-1930. 380. Bastrup, J., et al., Proteomic and Unbiased Post-Translational Modification Profiling of Amyloid Plaques and Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J Alzheimers Dis, 2020. 73(1): p. 393-411. 381. Liao, L., et al., Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem, 2004. 279(35): p. 37061-8. 382. Lutz, B.M. and J. Peng, Deep Profiling of the Aggregated Proteome in Alzheimer’s Disease: From Pathology to Disease Mechanisms. Proteomes, 2018. 6(4): p. 46. 383. Xiong, F., W. Ge, and C. Ma, Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimers Dement, 2019. 15(3): p. 429-440. 384. Drummond, E., et al., Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol, 2017. 133(6): p. 933-954. 385. Nijholt, D.A., C. Stingl, and T.M. Luider, Laser capture microdissection of fluorescently labeled amyloid plaques from Alzheimer’s disease brain tissue for mass spectrometric analysis. Methods Mol Biol, 2015. 1243: p. 165-73. 386. Rijal Upadhaya, A., et al., Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain, 2014. 137(Pt 3): p. 887-903. 387. Hu, Z.W., et al., Molecular structure of an N-terminal phosphorylated beta-amyloid fibril. Proc Natl Acad Sci U S A, 2019. 116(23): p. 11253-11258. 388. Michno, W., et al., Pyroglutamation of amyloid-betax-42 (Abetax-42) followed by Abeta1-40 deposition underlies plaque polymorphism in progressing Alzheimer’s disease pathology. J Biol Chem, 2019. 294(17): p. 6719-6732. 389. Roher, A.E., et al., Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem, 1993. 268(5): p. 3072-83. 390. Wildburger, N.C., et al., Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci Rep, 2017. 7(1): p. 9520. 391. Wisniewski, T., J. Ghiso, and B. Frangione, Biology of A beta amyloid in Alzheimer’s disease. Neurobiol Dis, 1997. 4(5): p. 313-28. 392. Miklossy, J., Bacterial Amyloid and DNA are Important Constituents of Senile Plaques: Further Evidence of the Spirochetal and Biofilm Nature of Senile Plaques. J Alzheimers Dis, 2016. 53(4): p. 1459-73. 393. Itzhaki, R.F., et al., Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol, 2020. 16(4): p. 193-197. 394. Itzhaki, R.F., et al., Microbes and Alzheimer’s Disease. J Alzheimers Dis, 2016. 51(4): p. 979-84. 395. Moir, R.D., R. Lathe, and R.E. Tanzi, The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement, 2018. 14(12): p. 1602-1614. 396. Lovheim, H., et al., Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement, 2015. 11(6): p. 593-9. 397. Readhead, B., et al., Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron, 2018. 99(1): p. 64-82 e7. 398. Sochocka, M., K. Zwolinska, and J. Leszek, The Infectious Etiology of Alzheimer’s Disease. Curr Neuropharmacol, 2017. 15(7): p. 996-1009. 399. Rizzo, R., Controversial role of herpesviruses in Alzheimer’s disease. PLoS Pathog, 2020. 16(6): p. e1008575. 400. Mawanda, F. and R. Wallace, Can infections cause Alzheimer’s disease? Epidemiol Rev, 2013. 35: p. 161-80. 401. Fulop, T., et al., Role of Microbes in the Development of Alzheimer’s Disease: State of the Art - An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet, 2018. 9: p. 362. 402. Fulop, T., et al., Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front Aging Neurosci, 2018. 10: p. 224. 403. Eimer, W.A., et al., Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron, 2018. 99(1): p. 56-63 e3. 404. Soscia, S.J., et al., The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One, 2010. 5(3): p. e9505. 405. Zhan, X., B. Stamova, and F.R. Sharp, Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front Aging Neurosci, 2018. 10: p. 42. 406. Harach, T., et al., Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep, 2017. 7: p. 41802. 407. Jucker, M. and L.C. Walker, Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci, 2018. 21(10): p. 1341-1349. 408. Jaunmuktane, Z. and S. Brandner, Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol Appl Neurobiol, 2019. 46(6): p. 522-545. 409. Ayers, J.I., N.A. Paras, and S.B. Prusiner, Expanding spectrum of prion diseases. Emerging Topics in Life Sciences, 2020: p. 1-13. 410. Prusiner, S.B., Development of the prion concept, in Prion Biology and Diseases, S.B. Prusiner, Editor. 2004, Cold Spring Harbor Laboratory Press: Cold Spring Harbor. p. 89-141. 411. Zabel, M.D. and C. Reid, A brief history of prions. Pathog Dis, 2015. 73(9): p. ftv087. 412. Prusiner, S.B., Prions. Proc Natl Acad Sci U S A, 1998. 95(23): p. 13363-83. 413. Friesen, M. and M. Meyer-Luehmann, Abeta Seeding as a Tool to Study Cerebral Amyloidosis and Associated Pathology. Front Mol Neurosci, 2019. 12: p. 233. 414. Morales, R., K. Callegari, and C. Soto, Prion-like features of misfolded Abeta and tau aggregates. Virus Res, 2015. 207: p. 106-12. 415. Watts, J.C. and S.B. Prusiner, beta-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. Cold Spring Harb Perspect Med, 2018. 8(5): p. a023507. 416. Asher, D.M., et al., Risk of Transmissibility From Neurodegenerative Disease-Associated Proteins: Experimental Knowns and Unknowns. J Neuropathol Exp Neurol, 2020. 79(11): p. 1141-1146. 417. Lauwers, E., et al., Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurology, 2020. 19: p. 872-878. 418. Brown, P., et al., Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis, 2012. 18(6): p. 901-7. 419. Will, R.G., Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull, 2003. 66: p. 255-65. 420. Jaunmuktane, Z., et al., Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature, 2015. 525(7568): p. 247-50. 421. Ritchie, D.L., et al., Amyloid-beta accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol, 2017. 134(2): p. 221-240. 422. Frontzek, K., et al., Amyloid-beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med Wkly, 2016. 146: p. w14287. 423. Hamaguchi, T., et al., Significant association of cadaveric dura mater grafting with subpial Abeta deposition and meningeal amyloid angiopathy. Acta Neuropathol, 2016. 132(2): p. 313-315. 424. Irwin, D.J., et al., Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol, 2013. 70(4): p. 462-8. 425. Kovacs, G.G., et al., Dura mater is a potential source of Abeta seeds. Acta Neuropathol, 2016. 131(6): p. 911-23. 426. Duyckaerts, C., et al., Neuropathology of iatrogenic Creutzfeldt-Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol, 2018. 135(2): p. 201-212. 427. Purro, S.A., et al., Transmission of amyloid-beta protein pathology from cadaveric pituitary growth hormone. Nature, 2018. 564(7736): p. 415-419. 428. Nakayama, H., et al., Cerebral amyloid angiopathy in an aged great spotted woodpecker (Picoides major). Neurobiol Aging, 1999. 20(1): p. 53-6. 429. Cork, L.C., et al., Neurofibrillary tangles and senile plaques in aged bears. J Neuropathol Exp Neurol, 1988. 47(6): p. 629-41. 430. Tekirian, T.L., et al., Carboxy terminal of beta-amyloid deposits in aged human, canine, and polar bear brains. Neurobiol Aging, 1996. 17(2): p. 249-57. 431. Uchida, K., et al., Senile plaques and other senile changes in the brain of an aged American black bear. Vet Pathol, 1995. 32(4): p. 412-4. 432. Cummings, B.J., et al., Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem, 1996. 66(1): p. 11-23. 433. Ishihara, T., et al., Immunohistochemical and immunoelectron microscopical characterization of cerebrovascular and senile plaque amyloid in aged dogs’ brains. Brain Res, 1991. 548(1-2): p. 196-205. 434. Uchida, K., et al., Immunohistochemical analysis of constituents of senile plaques and cerebro-vascular amyloid in aged dogs. J Vet Med Sci, 1992. 54(5): p. 1023-9. 435. Zeiss, C.J., Utility of spontaneous animal models of Alzheimer’s disease in preclinical efficacy studies. Cell Tissue Res, 2020. 380(2): p. 273-286. 436. Nakamura, S., et al., Senile plaques in very aged cats. Acta Neuropathol, 1996. 91(4): p. 437-9. 437. Nakamura, S., et al., Senile plaques in an aged two-humped (Bactrian) camel (Camelus bactrianus). Acta Neuropathol, 1995. 90(4): p. 415-8. 438. Roertgen, K.E., et al., A beta-associated cerebral angiopathy and senile plaques with neurofibrillary tangles and cerebral hemorrhage in an aged wolverine (Gulo gulo). Neurobiol Aging, 1996. 17(2): p. 243-7. 439. Heuer, E., et al., Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des, 2012. 18(8): p. 1159-69. 440. Jankowsky, J.L., et al., Rodent A beta modulates the solubility and distribution of amyloid deposits in transgenic mice. J Biol Chem, 2007. 282(31): p. 22707-20. 441. Steffen, J., et al., Expression of endogenous mouse APP modulates beta-amyloid deposition in hAPP-transgenic mice. Acta Neuropathol Commun, 2017. 5(1): p. 49. 442. Price, D.L., et al., Aged non-human primates: an animal model of age-associated neurodegenerative disease. Brain Pathol, 1991. 1(4): p. 287-96. 443. Selkoe, D.J., et al., Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer’s disease. Science, 1987. 235(4791): p. 873-7. 444. Walker, L.C. and L.C. Cork, The neurobiology of aging in nonhuman primates, in Alzheimer Disease, R.D. Terry, et al., Editors. 1999, Lippincott Williams and Wilkins: Philadelphia. p. 233-243. 445. Walker, L.C., Animal models of cerebral beta-amyloid angiopathy. Brain Res Brain Res Rev, 1997. 25(1): p. 70-84. 446. Jakel, L., et al., Animal models of cerebral amyloid angiopathy. Clin Sci (Lond), 2017. 131(19): p. 2469-2488. 447. Rosen, R.F., L.C. Walker, and H. Levine, 3rd, PIB binding in aged primate brain: enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol Aging, 2011. 32(2): p. 223-34. 448. Games, D., et al., Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature, 1995. 373(6514): p. 523-7. 449. Hsiao, K., et al., Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science, 1996. 274(5284): p. 99-102. 450. Sturchler-Pierrat, C., et al., Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A, 1997. 94(24): p. 13287-92. 451. Duyckaerts, C., M.C. Potier, and B. Delatour, Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol, 2008. 115(1): p. 5-38. 452. Jucker, M., The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med, 2010. 16(11): p. 1210-4. 453. Myers, A. and P. McGonigle, Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr Protoc Neurosci, 2019. 89(1): p. e81. 454. Greeve, I., et al., Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci, 2004. 24(16): p. 3899-906. 455. Iijima-Ando, K. and K. Iijima, Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struct Funct, 2010. 214(2-3): p. 245-62. 456. Jeon, Y., et al., Genetic Dissection of Alzheimer’s Disease Using Drosophila Models. Int J Mol Sci, 2020. 21(3): p. 884. 457. Link, C.D., C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol, 2006. 41(10): p. 1007-13. 458. Link, C.D., et al., Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging, 2001. 22(2): p. 217-26. 459. Dawson, T.M., T.E. Golde, and C. Lagier-Tourenne, Animal models of neurodegenerative diseases. Nat Neurosci, 2018. 21(10): p. 1370-1379. 460. Gotz, J. and L.M. Ittner, Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci, 2008. 9(7): p. 532-44. 461. Rockenstein, E., L. Crews, and E. Masliah, Transgenic animal models of neurodegenerative diseases and their application to treatment development. Adv Drug Deliv Rev, 2007. 59(11): p. 1093-102. 462. Gotz, J., L.G. Bodea, and M. Goedert, Rodent models for Alzheimer disease. Nat Rev Neurosci, 2018. 19(10): p. 583-598. 463. Kitazawa, M., R. Medeiros, and F.M. Laferla, Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des, 2012. 18(8): p. 1131-47. 464. Walker, L.C., et al., Labeling of cerebral amyloid in vivo with a monoclonal antibody. J Neuropathol Exp Neurol, 1994. 53(4): p. 377-83. 465. Schenk, D., et al., Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature, 1999. 400(6740): p. 173-7. 466. Braakman, N., et al., Longitudinal assessment of Alzheimer’s beta-amyloid plaque development in transgenic mice monitored by in vivo magnetic resonance microimaging. J Magn Reson Imaging, 2006. 24(3): p. 530-6. 467. Christie, R.H., et al., Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J Neurosci, 2001. 21(3): p. 858-64. 468. Dong, J., et al., Multiphoton in vivo imaging of amyloid in animal models of Alzheimer’s disease. Neuropharmacology, 2010. 59(4-5): p. 268-75. 469. Rominger, A., et al., Longitudinal assessment of cerebral beta-amyloid deposition in mice overexpressing Swedish mutant beta-amyloid precursor protein using 18F-florbetaben PET. J Nucl Med, 2013. 54(7): p. 1127-34. 470. Prada, C.M., et al., Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci, 2007. 27(8): p. 1973-80. 471. Okano, H. and N. Kishi, Investigation of brain science and neurological/psychiatric disorders using genetically modified non-human primates. Curr Opin Neurobiol, 2018. 50: p. 1-6. 472. Seita, Y., et al., Generation of Transgenic Cynomolgus Monkeys Overexpressing the Gene for Amyloid-beta Precursor Protein. J Alzheimers Dis, 2020. 75(1): p. 45-60. 473. Vermunt, L., et al., Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement, 2019. 15(7): p. 888-898. 474. Selkoe, D.J., Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med, 2011. 17(9): p. 1060-5. 475. Selkoe, D.J., Editorial: A Is for Amyloid. J Prev Alzheimers Dis, 2020. 7(3): p. 140-141. 476. Tolar, M., et al., Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther, 2020. 12(1): p. 95. 477. Uhlmann, R.E., et al., Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nature Neuroscience, 2020. (in press). 478. Holmes, C., et al., Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet, 2008. 372(9634): p. 216-23. 479. Lozupone, M., et al., Anti-amyloid-beta protein agents for the treatment of Alzheimer’s disease: an update on emerging drugs. Expert Opin Emerg Drugs, 2020: p. 1-17. 480. Clavaguera, F., C. Duyckaerts, and S. Haik, Prion-like properties of Tau assemblies. Curr Opin Neurobiol, 2020. 61: p. 49-57. 481. Holmes, B.B. and M.I. Diamond, Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem, 2014. 289(29): p. 19855-61. 482. Klein, G., et al., Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther, 2019. 11(1): p. 101. 483. Sevigny, J., et al., The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature, 2016. 537(7618): p. 50-6. 484. Selkoe, D.J. and J. Hardy, The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 2016. 8(6): p. 595-608. 485. Angelucci, F., et al., Antibiotics, gut microbiota, and Alzheimer’s disease. J Neuroinflammation, 2019. 16(1): p. 108. 486. Sureda, A., et al., Oral microbiota and Alzheimer’s disease: Do all roads lead to Rome? Pharmacol Res, 2020. 151: p. 104582. 487. Lemere, C.A., Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener, 2013. 8: p. 36. 488. van Dyck, C.H., Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry, 2018. 83(4): p. 311-319. 489. Voytyuk, I., B. De Strooper, and L. Chavez-Gutierrez, Modulation of gamma- and beta-Secretases as Early Prevention Against Alzheimer’s Disease. Biol Psychiatry, 2018. 83(4): p. 320-327. 490. Wisniewski, T. and F. Goni, Immunotherapeutic approaches for Alzheimer’s disease. Neuron, 2015. 85(6): p. 1162-76. 491. Plotkin, S.S. and N.R. Cashman, Passive immunotherapies targeting Abeta and tau in Alzheimer’s disease. Neurobiol Dis, 2020. 144: p. 105010. 492. Kwon, S., et al., Immunotherapies for Aging-Related Neurodegenerative Diseases-Emerging Perspectives and New Targets. Neurotherapeutics, 2020. p. 1-20. 493. Yu, Y.J. and R.J. Watts, Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics, 2013. 10(3): p. 459-72. 494. Bennett, C.F., A.R. Krainer, and D.W. Cleveland, Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu Rev Neurosci, 2019. 42: p. 385-406. 495. Chakravarthy, M., et al., Nucleic Acid-Based Theranostics for Tackling Alzheimer’s Disease. Theranostics, 2017. 7(16): p. 3933-3947. 496. Kim, K.S., et al., Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun, 1988. 2: p. 121-130. 497. Potempska, A., et al., Quantification of sub-femtomole amounts of Alzheimer amyloid beta peptides. Amyloid, 1999. 6(1): p. 14-21. 498. Horikoshi, Y., et al., Development of Abeta terminal end-specific antibodies and sensitive ELISA for Abeta variant. Biochem Biophys Res Commun, 2004. 319(3): p. 733-7. 499. Duff, K., et al., Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis, 2000. 7(2): p. 87-98. 500. Jicha, G.A., et al., Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res, 1997. 48(2): p. 128-32. 501. Imai, Y., et al., A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun, 1996. 224(3): p. 855-62. 502. Ito, D., et al., Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res, 1998. 57(1): p. 1-9. 503. Lichtenberg-Kraag, B., et al., Phosphorylation-dependent epitopes of neurofilament antibodies on tau protein and relationship with Alzheimer tau. Proc Natl Acad Sci U S A, 1992. 89(12): p. 5384-8. 504. Goldstein, M.E., L.A. Sternberger, and N.H. Sternberger, Varying degrees of phosphorylation determine microheterogeneity of the heavy neurofilament polypeptide (Nf-H). J Neuroimmunol, 1987. 14(2): p. 135-48. 505. Sternberger, L.A. and N.H. Sternberger, Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci U S A, 1983. 80(19): p. 6126-30. 506. Naoumenko, J. and I. Feigin, A stable silver solution for axon staining in paraffin sections. J Neuropathol Exp Neurol, 1967. 26(4): p. 669-673. 507. Campbell, S.K., R.C. Switzer III, and T.L. Martin, Alzheimer’s plaques and tangles: a controlled and enhanced silver staining method. Society for Neuroscience Abstracts, 1987. 13: p. 687. 508. Cowe, A., Der gliöse Anteil der senilen Plaques. Zeitschrift für die gesamte Neurologie und Psychiatrie, 1915. 29(1): p. 92-96. 509. McMenemey, W.H., Alzheimer’s disease: Problems concerning its concept and nature. Acta Neurologica Scandinavica, 1963. 39: p. 369-380. 510. DeFelipe, J., Cajal’s Butterfiles of the Soul. 2010, Oxford University Press: New York. 511. Alafuzoff et al., Inter-laboratory comparison of neuropathological assessments of beta-amyloid protein: a study of the BrainNet Europe consortium. Acta Neuropathologica, 2008. 115[5]: p. 533-546 512. Stonebarger et al., Amyloidosis increase is not attenuated by long-term calorie restriction or related to neuron density in the prefrontal cortex of extremely aged rhesus macaques. Geroscience, 2020. doi: 10.1007/s11357-020-00259-0.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |