|
|
|
Free Neuropathology 1:23 (2020) |
|
Original Paper |
|
Neuronal Transcriptome from C9orf72 Repeat Expanded Human Tissue is Associated with Loss of C9orf72 Function |
|
Elaine Y. Liu1, Jenny Russ1, Edward B. Lee1 |
|
1 Translational Neuropathology Research Laboratory, University of Pennsylvania, Philadelphia, PA, USA |
|
Corresponding author: |
|
Submitted: 20 July 2020 Accepted: 13 August 2020 Copyedited by: Lauren Walker Published: 21 August 2020 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Dementia, Transcriptome, Frontotemporal, Amyotrophic lateral sclerosis, C9orf72, TDP-43, FACS, RNA-seq, Repeat expansion, DENN, Frontotemporal lobar degeneration, Frontotemporal dementia |
|
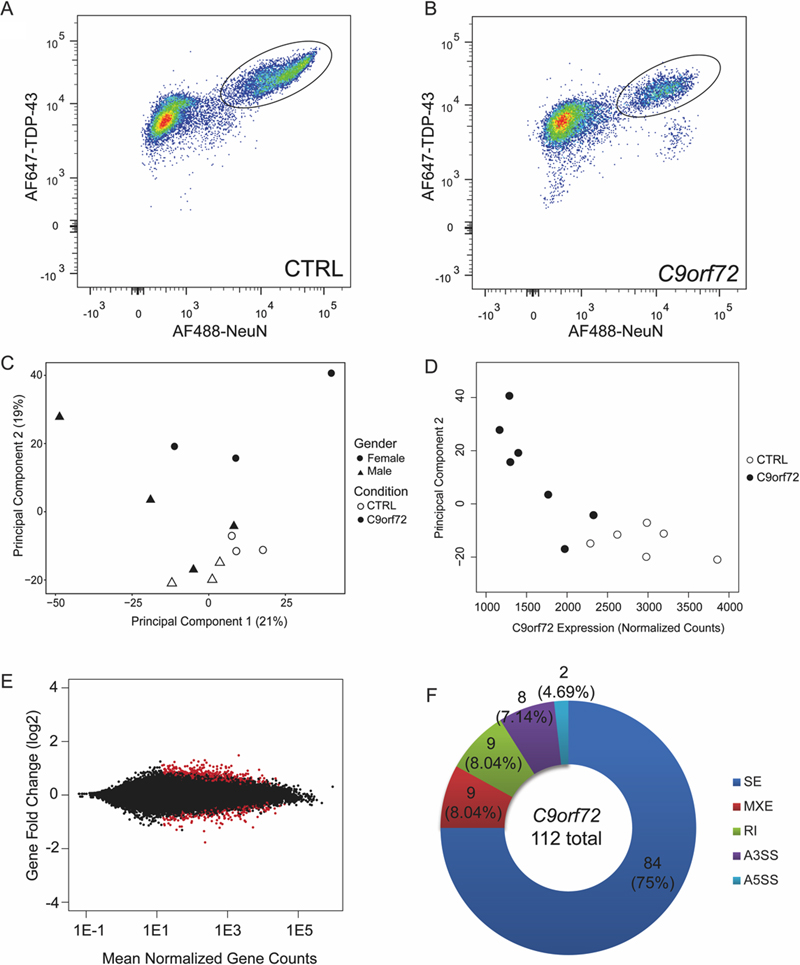
Abstract A hexanucleotide G4C2 repeat expansion in C9orf72 is the most common genetic cause of familial and sporadic cases of amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD). The mutation is associated with a reduction of C9orf72 protein and accumulation of toxic RNA and dipeptide repeat aggregates. The accumulation of toxic RNA has been proposed to sequester RNA binding proteins thereby altering RNA processing, consistent with previous transcriptome studies that have shown that the C9orf72 repeat expansion is linked to abundant splicing alterations and transcriptome changes. Here, we used a subcellular fractionation method and FACS to enrich for neuronal nuclei from C9orf72 repeat expanded post-mortem human ALS/FTD brains, and to remove neuronal nuclei with TDP-43 pathology which are observed in nearly all symptomatic C9orf72 repeat expanded cases. We show that the C9orf72 expansion is associated with relatively mild gene expression changes. Dysregulated genes were enriched for vesicle transport pathways, which is consistent with the known functions of C9orf72 protein. Further analysis suggests that the C9orf72 transcriptome is not driven by toxic RNA but is rather shaped by the depletion of pathologic TDP-43 nuclei and the loss of C9orf72 expression. These findings argue against RNA binding protein sequestration in neurons as a major contributor to C9orf72 mediated toxicity. Introduction Amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) are fatal neurodegenerative diseases with overlapping clinical, pathologic and genetic features. ALS is a motor neuron disease that primarily affects the upper and lower motor neurons in the motor cortex and spinal cord, respectively, whereas FTD affects the frontal and temporal lobes, thereby affecting cognition. The most common genetic cause of familial and sporadic cases of ALS and FTD was identified as a hexanucleotide G4C2 repeat expansion found in the gene C9orf72 (1, 2). The repeat expansion is found in the first intron of C9orf72 and can be transcribed into pre-mRNA that contains the repeat. C9orf72 protein has been predicted to be a DENN Rab GTPase, and shown to be involved in vesicular trafficking (3-8). Three potential mechanisms of toxicity have been implicated in contributing to disease: haploinsufficiency due to reduction of C9orf72 protein; RNA foci and titration of RNA binding proteins; and dipeptide repeat protein aggregates (reviewed in (9)). It is currently unclear which mechanism contributes most to toxicity. Gain of toxic function via RNA foci or dipeptide repeat proteins have been seen in cellular and animal models. Indeed, several RNA binding proteins have been shown to colocalize with repeat containing RNA within RNA foci, suggesting that RNA binding protein titration may be mediating toxicity (10-12). Models overexpressing dipeptide repeat proteins and/or mutant RNA in both cell and animal systems have shown that the repeat expansion and dipeptide proteins may confer toxicity via a variety of pathways including nucleolar stress, nucleocytoplasmic transport defects, heterochromatin abnormalities, and DNA damage (13-32). However, RNA foci and dipeptide repeat proteins do not correlate with neurodegeneration in human tissues and are not toxic in some experimental models (13-16, 33-36), raising questions about the relevance of whether these pathologies contribute to disease. The loss of C9orf72 protein has been substantiated by the fact that C9orf72 expansion carriers have reduced C9orf72 mRNA and protein expression (1, 2, 37, 38). Thus, it is possible that loss of C9orf72 protein may contribute to disease. However, knockout mouse models of C9orf72 do not show neuronal defects but, rather, exhibit an inflammatory phenotype (39-41), although reduction of C9orf72 protein appears to exacerbate phenotypes in mice overexpressing mutant C9orf72 RNA (42). We have shown that a subset of C9orf72 expansion carriers exhibit C9orf72 promoter methylation, and this is associated with reduced C9orf72 associated pathology, later age at death in FTD patients, slower rates of cerebral atrophy, and improved cognitive ability (43-45). Moreover, the gnomAD human genetics database indicates that missense and loss of function C9orf72 mutations are tolerated and present in control populations (46). Consequently, the pathogenic role of the loss of C9orf72 protein in ALS/FTD is not entirely clear, and overall it remains unclear which mechanism of toxicity contributes most to disease. Efforts to better understand these mechanisms include transcriptomic studies to determine whether RNA dysregulation can contribute to neurodegeneration. Indeed, frontal cortical and motor neuron transcriptome analysis from ALS patients showed splicing changes, dysregulation of RNA processing pathways, and alternative polyadenylation insporadic and C9orf72 expansion carriers (47-49). More recently, a large transcriptome study of C9orf72 cases found changes in genes linked to vesicular transport (8). These studies are sometimes complicated by the complex cellular composition of brain tissue which includes neuronal subtypes, oligodendrocytes, microglia, astrocytes, vascular cells and other cell types. Laser capture microdissected tissue studies have been very useful (50) but is labor intensive and often is technically difficult due to relatively low yield and preprocessing steps that can affect RNA quality (51). Single cell RNA sequencing methods have recently addressed the issue of complex cellular mixtures within tissue but suffer from low coverage and data sparsity. We have previously reported the use of fluorescence activated cell sorting (FACS) to identify transcriptome signatures in neuronal nuclei without TDP-43 from post-mortem brain in ALS/FTD patients (52). Here, we have used the same method to determine the role of the C9orf72 repeat expansion in post-mortem neurons from ALS/FTD patients. Compared to the profound transcriptome changes associated with the loss of normal nuclear TDP-43 protein (52), we found that the C9orf72 mutation was associated with relatively mild gene expression changes and mild splicing changes. Moreover, the molecular signature is largely associated with a loss of C9orf72 function, and not a gain of toxic RNA or dipeptide repeat aggregates. Materials and Methods Clinical and pathologic assessment Human autopsy tissue was obtained from the University of Pennsylvania Center for Neurodegenerative Disease Research Brain Bank as described (53). Informed consent from next of kin was obtained for every case. Isolation was performed exactly as previously described (52). Briefly, mid-frontal neocortex of all cases were dounce homogenized using pestil B (Kimble Chase) in 0.25M sucrose and adjusted to final molarity of 1.6M sucrose in TKM. The homogenate was spun on a 1.8M sucrose cushion on the Beckman Coulter XPN-80 ultracentrifuge at 40,000g for 40 minutes at 4°C (Beckman Coulter Inc, Indianapolis, IN, USA). Isolated nuclei were stained with Alexa Fluor 647 conjugated to 2089 (rabbit polyclonal C-terminal anti-TDP-43 antibody, Center for Neurodegenerative Disease Research, University of Pennsylvania), Alexa Fluor 488 conjugated NeuN (EMD Millipore, Billerica, MA, USA), and DAPI (Invitrogen, Carlsbad, CA, USA). Alexa Fluor 647 was conjugated to 2089 according to the APEX Alexa Fluor 647 Antibody labeling kit protocol (Thermo Fisher Scientific, Waltham, MA, USA). Stained nuclei were sorted for single cells containing TDP-43 and NeuN on the BD FACSAria II (BD Biosciences, San Jose, CA, USA) at 20 psi on 100µm nozzle. RNA Isolation and RNA-seq Library Generation Isolation and library generation was done as previously described (52). Briefly, RNA was extracted using the standard protocol within the AllPrep DNA/RNA Micro kit (Qiagen, Germantown, MD, USA). RNA quality from sorted nuclear RNA was determined based on Bioanalyzer Picochip analysis (Agilent, Santa Clara, CA, USA). Isolated RNA was amplified, made into cDNA, sheared, and libraries were made. The library was quantified using the Qubit dsDNA kit (Invitrogen) and Kapa library quantification kit (KapaBiosystems, Boston, MA). cDNA libraries were pooled, clustered on the cBot and subject to 100 or 125 base pairs paired end reads on the HiSeq 2000 or 2500 (Illumina, San Diego, CA, USA). Pre-processing, mapping and filtering of RNA-seq data RNA-seq analysis was done as previously described (52). Briefly, raw sequencing reads were demultiplexed through the UPenn Functional Genomics Core and analyzed for quality control using FastQC. Reads were mapped to the human genome (GRCh38, GENCODE release 22) using STAR and only uniquely mapping reads were selected for further analysis. Ribosomal and mitochondrial reads were removed and SAM files were converted to BAM files using samtools view and BAM files were sorted by coordinate with samtools sort. Creation of non-overlapping gene, exon and intron annotations Annotations were generated as previously described (52). Briefly, annotations were based on the comprehensive gene annotation file of the GENCODE Release 22 (GRCh38.p2). GTF file was loaded into R (Version 3.2.2; R Core Team (2015): “R: A Language and Environment for Statistical Computing”, R Foundation for Statistical Computing, Vienna, Austria) and converted into a TranscriptDb object. From the TranscriptDb object, all annotated Ensembl genes and their exons were pulled out using exonsBy (by=”gene”) and Ensembl gene IDs were replaced by official gene symbols with biomaRt (Version 2.26.1). Genes and introns were defined as previously described in the above chapter. Regions shared by overlapping genes were removed to count reads that map to one gene or to genic elements (exon or intron) from one gene. Exons and introns annotations were used for subsequent analyses in the R package DEXSeq (1.16.10). Differential gene and genic element expression analysis The genomic annotation used for read counting was done as previously described (52). The mapped, filtered RNA sequencing reads were counted using a custom R script including the R packages Rsamtools (1.22.0), GenomicFeatures (1.22.8) and GenomicAlignments (1.6.3). Briefly, sorted BAM files were loaded into R (3.2.2) and the number of reads mapping to genes, exons or introns, was computed. Genes were analyzed for differential expression using the R package DESeq2 (1.10.1). Analysis was used to determine differences between nuclei from both C9orf72 expansion cases and non-diseased controls (option design=~experiment in DESeqDataSetFromMatrix tool where experiment is the subject id). Sex effects were removed with the R package sva (3.18.0) according to the DESeq2 vignette. Manual annotation of gene descriptions was performed using gene descriptions on NCBI. Differential genic element expression analysis was done using DEXSeq (1.16.10). Briefly, the sorted BAM files were used to count the number of reads mapping to exons and introns using the exon and intron annotation generated above and computed using findOverlaps and countSubjectHits. Reads mapping to exon-intron junctions were excluded. Sex was controlled for by including it in the linear model used in the analysis by adding the term “sex:exon”. The following linear models were used full model = ~sample + exon + sex:exon + experiment:exon and reduced model = ~sample + exon + sex:exon + experiment:exon and reduced model = ~sample + exon + sex:exon, where sample is the sample id and experiment the subject id. In addition, dispersions were estimated using the tool estimateDispersions with option fitType=’local’. Changes in expression were significant if Bonferroni-Hochberg multiple testing adjusted p-values were less than 0.05. Alternative splicing analysis Splicing analysis was performed as previously described (52). Briefly, all FASTQ files were trimmed to 100bp, aligned to GRCh38 using STAR; ribosomal and mitochondrial mapped reads were removed. SAM files were converted to sorted BAM files and rMATS.3.0.9 was run using default parameters with the following options (-t paired –len 100 –c 0.05 – analysis U). Significant alternative splicing events were used if Bonferroni-Hochberg multiple testing adjusted p-values were less than 0.05. RNA binding protein CLIP analysis CLIP analysis was done as previously described (52) using published TDP-43 iCLIP data from SH-SY5Y cells (54) and hnRNP A, A2B1, F, M, U CLIP data from HEK293 cells (55). Briefly, hg18 Bowtie files were converted to FASTQ files, and aligned to hg38. PIPECLIP was run using the python script. Using R, the “GenomicRanges” package with ‘findOverlaps’ option was used to determine which bins had RNA binding protein sites. Chi-square analysis was done to determine whether there was a significant enrichment of bins with RNA binding protein sites within the significantly differentially used bins linked to the C9orf72 mutation. Principal Component Analysis Principal component analysis was done using the log transformed read count dataset in R with the ‘stats’ package and ‘prcomp’ function. The coordinates were retrieved and used to plot principal component 2 (PC2) vs principal component 1. To determine the relationship between PC2 vs C9orf72 expression, the coordinates of PC2 for each sample was plotted against the normalized C9orf72 expression calculated from DESeq2 from each sample. Pearson’s correlation was performed to determine the correlation between PC2 and C9orf72 expression. Gene ontology analysis of the top 1% of all genes that contribute to PC2 was done using Webgestaldt (56) with the overrepresentation enrichment analysis using the ‘geneontology’ functional database with a FDR corrected significance level < 0.05. Methylation Correlation Methylation levels for the C9orf72 promoter in patients were determined as previously described (45). Briefly, genomic DNA from the cerebellum was extracted using the Qiagen DNeasy Blood and Tissue kit and subject to overnight digestion with HhaI and HaeIII (double-digested) or just HaeIII (mock) alone. A small aliquot of DNA was amplified using primers flanking the HhaI cutsite within the C9orf72 promoter region using 2x FastStart SYBR Green Master (Roche) on the ABI StepOnePlus machine. The difference in cycles to threshold amplification between double and mock digested DNA was calculated as methylation values. Spearman’s correlation was calculated using gene counts for each gene and methylation values for each C9orf72 mutation case. R package ‘lsr’ with ‘correlate’ function (with options corr.method=”spearman” and p.adjust.method=”fdr”) using the Spearman’s correlation and FDR adjusted p-values was used. Only correlations from genes with HUGO gene symbols were calculated and plotted against gene fold change for each gene that was calculated by DESeq2. This was done using both significantly differentially expressed genes linked to the C9orf72 mutation (DESeq2 FDR p-value < 0.05) and genome-wide using all expressed genes. Results Fluorescence Activated Cell Sorting and RNA Sequencing of Sorted Neuronal Nuclei The C9orf72 mutation leads to TDP-43 pathology which includes the formation of neuronal cytoplasmic inclusions and the loss of normal physiologic nuclear TDP-43 protein. We have previously shown that the loss of normal nuclear TDP-43 protein has large effects on the nuclear transcriptome (52). To determine whether the C9orf72 mutation leads to transcriptomic alterations independent of TDP-43 pathology, we isolated neuronal nuclei with intact TDP-43 expression. Mid-frontal neocortex from 7 post-mortem C9orf72 expansion carriers and 6 neurologically normal controls were used to isolate nuclei for transcriptome-wide analysis. The autopsy cohort characteristics can be found in Table 1. Nuclei were immunostained for NeuN and TDP-43 and subjected to FAC sorting to isolate NeuN positive, TDP-43 positive nuclei from controls (circled, Fig 1A) and C9orf72 expansion cases (circled, Fig 1B). RNA from equal numbers of TDP-43 positive neuronal nuclei (35,000-100,000) was extracted and amplified to generate barcoded cDNA libraries for 100 or 125bp paired end sequencing on the Illumina HiSeq 2000/2500. Sequences were mapped to the human genome (Gencode GRCh38) using STAR algorithms (version 2.2.4). There were 1.8 billion reads of which 1.065 billion reads mapped uniquely between the 13 libraries, with an average of 90 million uniquely mapped reads per library. These reads were filtered to remove ribosomal and mitochondrial reads and the resulting reads were used for downstream analysis to evaluate differential expression of genes, genic elements and alternative splicing.
Table 1: Autopsy cohort characteristics. ALS = amyotrophic lateral sclerosis; FTLD = frontotemporal lobar degeneration with TDP-43 inclusions; MND = motor neuron disease; NL = normal; PMI = post-mortem interval; RIN = RNA integrity number
Fig 1: C9orf72 repeat expansion contributes to relatively mild transcriptome changes. Flow cytometry plots of control (A) and C9orf72 expansion cases (B) with TDP-43 positive neuronal nuclei circled as collected samples. (C) Principal component analysis using all expressed genes where shape denotes sex and color denotes condition. (D) Principal component 2 strongly correlates with C9orf72 expression (Pearson’s r = -0.8205; p=0.0006). (E) MA plot showing gene fold change as a function of mean normalized gene counts with red dots being significantly differentially expressed genes. (F) Pie chart distribution of all alternative splicing events between C9orf72 expansion carriers and non-diseased controls. SE=skipped exons, MXE=mutually exclusive exons, RI=retained intron, A3SS=alternative 3’ splice site, A5SS=alternative 5’ splice site. Functional transcriptomic alterations associated with the C9orf72 hexanucleotide repeat expansion To verify that the variation within the dataset is linked to the mutation status, principal component analysis (PCA) was performed using the read counts of the entire dataset (Fig 1C). Principal component #1 (PC1) explained 21% of the variation whereas PC2 explained 19% of the variation (Fig 1C). Based on the clustering of the dataset, sex explains the variation in PC1 while the C9orf72 repeat expansion explains the variation in PC2. Indeed, PC2 strongly correlated with C9orf72 mRNA expression (Pearson’s r=-0.8205; p=0.0006), suggesting that C9orf72 expression is the major underlying biological variable that contributes to the variation in PC2 (Fig 1D). To identify whether there were enriched pathways within the genes that contribute most to PC2, gene ontology analysis was performed using the top 1% of all expressed genes (318 genes) that contribute to PC2. One pathway that is of interest is the cytoplasmic vesicle part or cytoplasmic vesicle membrane (Table 2) and is consistent with the functional role of C9orf72 as a vesicle trafficking protein and Rab GTPase (3, 5-7).
Table 2: Gene ontology (GO) of genes related to top 1% of all genes that contribute to variation in PC2. Previous studies have found that the G4C2 RNA foci may colocalize with different RNA binding proteins (RBPs), suggesting that sequestration of these RBPs can result in aberrant RNA processing (10-12). Differential gene expression analysis showed that there are 323 significantly differentially expressed genes with 202 upregulated and 121 downregulated genes linked to the C9orf72 mutation (Fig 1E, supplemental table 1). Similar to a previous transcriptome analysis on C9orf72 expansion carriers on the frontal cortex, there were more upregulated genes than downregulated genes within the differentially expressed genes (49). Notably, the number of differentially expressed genes linked the C9orf72 repeat expansion was relatively mild compared to the massive transcriptome-wide alterations we previously described associated with TDP-43 pathology in these same cases (5,576 significantly differentially expressed genes, described in (52)). Further annotation shows that these genes are generally involved in synaptic vesicle fusion or vesicle formation (Table 3). Additional genes related to vesicle transport and endosomal trafficking are also dysregulated. Interestingly, nine out of these 11 genes are upregulated. Additional genes that were upregulated were involved in protein aggregation. DNAJB2 is almost exclusively expressed in neurons and has been shown to resolve TDP-43 aggregates by interacting with heat shock protein 70 (57). MGRN1 has been shown to confer cytoprotective effects in an ALS mouse model and can suppress chaperone associated misfolded protein aggregation and toxicity (58). Given that C9orf72 expansion cases exhibit protein aggregates in the form of dipeptide repeat protein and TDP-43 inclusions, it is possible that these genes are upregulated in response to these aggregates. Lastly, there were also genes that are involved in DNA repair (FAAP20, FAM175A, FANCB, HMGN1) (59-62) and response to DNA damage (KDM4B and KIN) (63-65). Annotation of these genes reveal multiple themes relevant to C9orf72 including synaptic vesicle formation, endosomal trafficking, chaperone associated protein aggregation, and DNA damage (Table 3). C9orf72 mutation is associated with mild splicing alterations Previous studies have shown that there are abundant alternative splicing changes that are linked to the C9orf72 mutation (49). The finding of RNA binding proteins that colocalize with RNA foci also support the idea that RNA binding protein sequestration and subsequent splicing alterations may contribute to mutant C9orf72 toxicity. To evaluate splicing changes associated with the C9orf72 mutation, rMATS was used to align junction reads to annotated junctions from GRCh38 assembly. Within our dataset, there were a total of 112 events that were significantly alternatively spliced which included 84 skipped exons (SE), 9 mutually exclusive exons (MXE), 9 retained introns (RI), 8 alternative 3’ splice sites (A3SS), and 2 alternative 5’ splice site (A5SS) events (Fig 1F, supplemental table 2). These events affected 111 genes, of which only 3 were also significantly differentially expressed. Gene ontology analysis of 111 genes did not result in any significant enriched pathways. Therefore, we observed relatively mild splicing changes associated with the C9orf72 mutation. This may be due in part attributable to technical differences where the use of nuclear RNA in our study likely reduces sensitivity in terms of identifying splicing alterations.
Table 3: Annotation of significantly differentially expressed genes with general terms relevant to C9orf72 mutation. FDR = false discovery rate.
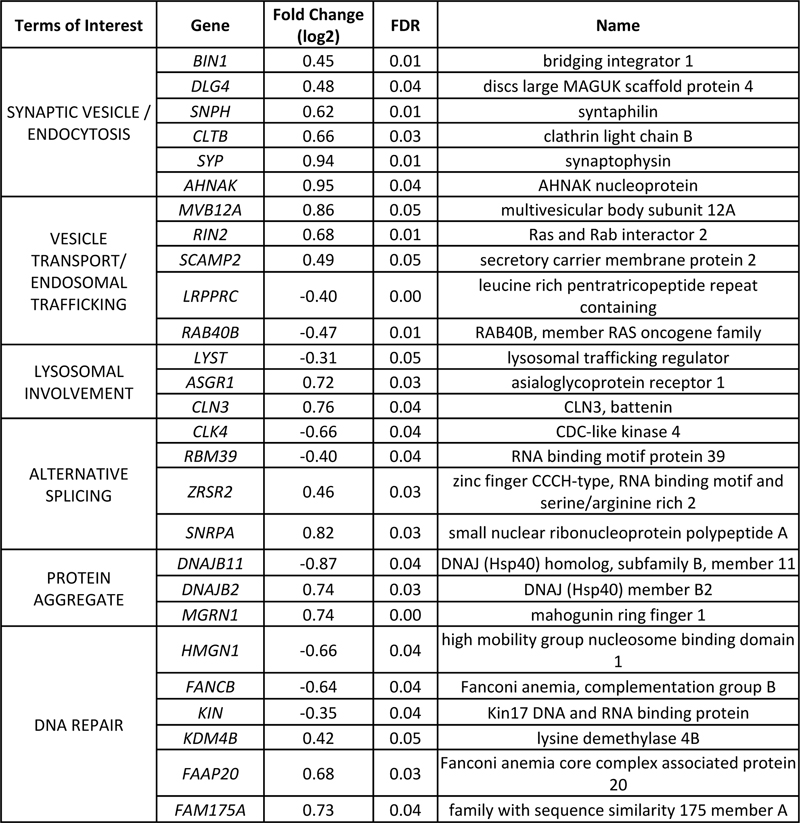
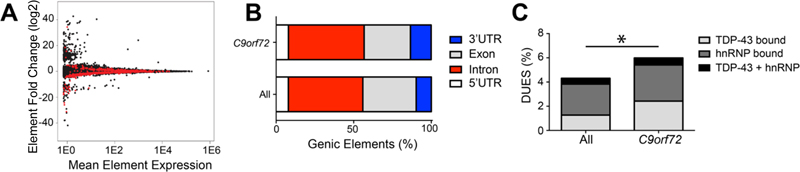
Fig 2: The C9orf72 mutation is associated with differential usage of genic elements with RNA binding protein sites. (A) MA plot of DEXSeq differentially used genic elements with red being significantly differentially used elements. (B) Genic elements (5’ UTR, exon, intron, 3’ UTR) distribution associated with C9orf72 mutation and all expressed bins. (c) Differentially used elements that are bound by TDP-43 and hnRNP (χ2=5.617; p=0.0178). C9orf72 mutation is associated with differentially used 3’ UTRs Altered RNA binding protein function has been proposed to contribute to C9orf72 toxicity. RNA binding proteins can bind to different genic elements including exons, introns and untranslated regions (UTR). If RBP activity is altered via titration of these proteins, it is possible the regions that may be bound by RBPs are also differentially expressed. Thus, DEXSeq was used to determine whether the presence of the repeat expansion resulted in differential usage of genic elements. There were a total of 865 significantly differentially used elements linked to the repeat expansion affecting 791 genes. Specifically, there were 610 significantly downregulated elements and 255 significantly upregulated elements (Fig 2A). When the elements were annotated as either 5’ UTR, exon, intron or 3’ UTR, there was a significant enrichment of differentially used 3’ UTRs due to the C9orf72 mutation (13.35% vs 9.78%, χ2=12.50, p=0.0004) (Fig 2B). Given the role of RBPs in binding to 3’ UTRs, we hypothesized that the significantly differentially used elements were enriched for RBP binding sites. Thus, hnRNP binding sites, including that of TDP-43 and other hnRNPs were identified using PIPE-CLIP algorithms applied to published data sets (54, 55, 66). Indeed, there was an enrichment of differentially used genic elements that contain TDP-43 and hnRNP binding sites (χ2=5.617, p=0.0178, Fig 2C). Thus, the C9orf72 expansion is associated with some differentially used genic elements with RBP binding sites. Transcriptomic signature due to depletion of TDP-43 pathologic nuclei Given that neurons with TDP-43 inclusions have died during the course of disease and our experimental design includes removal of neurons with TDP-43 pathology via FAC sorting before RNA sequencing, we next considered the possibility that the transcriptomic alterations associated with the C9orf72 mutation in this dataset would in part reflect the depletion of neurons vulnerable to TDP-43 proteinopathy. Indeed, within the neocortex, TDP-43 proteinopathy often preferentially affect neurons in superficial cortical laminae (52, 67). As shown in Fig 3A, neurons with TDP-43 pathology including loss of nuclear TDP-43 (white) are depleted in C9orf72 expansion cases, while control brains have the normal complement of neurons (blue, Fig 3B). As a result, we hypothesized that the depletion of diseased nuclei would contribute to the transcriptomic alterations associated with the C9orf72 mutation. Thus, we expected that common genes that were downregulated in C9orf72 expansion cases would be upregulated in nuclei without TDP-43 and vice versa. Indeed, among the 118 common significant differentially expressed genes linked to TDP-43 loss (52) and the C9orf72 mutation, a majority of them were followed this pattern (Fig 3C, supplemental table 3). A linear regression analysis showed there was a negative correlation between the gene fold change linked to TDP-43 loss and the gene fold change linked to the C9orf72 mutation (Pearson’s r=-0.5708; p<0.0001). Furthermore, this relationship was extended transcriptome-wide where we observed a negative correlation in all commonly expressed genes derived from both data sets (Fig 3D). For example, IGSF11 and PVRL3 expression, preferentially observed in superficial neocortical layers and therefore increased in neuronal nuclei without TDP-43 protein (52), is significantly reduced in the C9orf72 transcriptome. Conversely, SYT6 and COL6A1 expression, preferentially observed in deep neocortical layers and therefore decreased in neuronal nuclei without TDP-43 protein (52), is significantly increased in the C9orf72 transcriptome. This suggested that depletion of the diseased nuclei in C9orf72 expansion cases may explain some of the transcriptomic alterations observed in C9orf72 expansion cases.
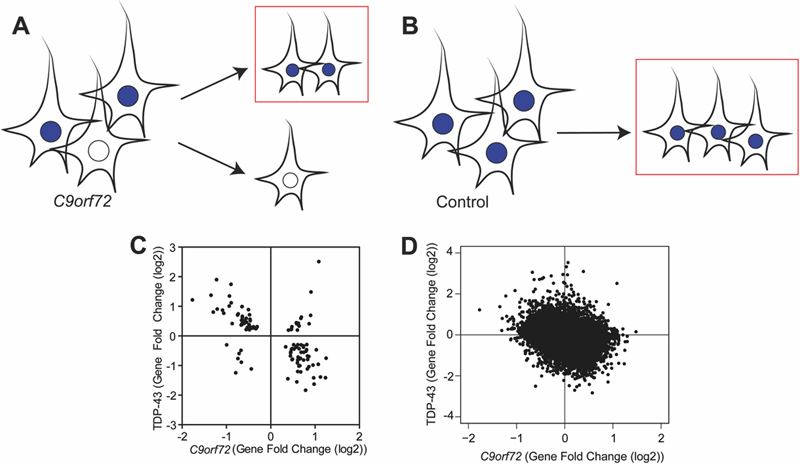
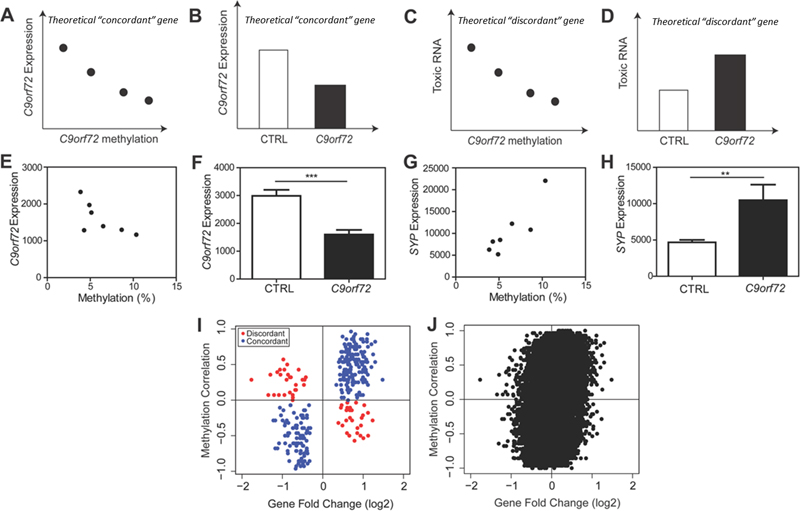
Fig 3: C9orf72 transcriptome is linked to depletion of TDP-43 pathologic nuclei. (A) In C9orf72 expansion cases, there are nuclei with TDP-43 (blue) and those without TDP-43 (white). For our transcriptome analysis, only nuclei with TDP-43 were collected (red box), thereby depleting nuclei without TDP-43. (B) In non-diseased controls, nuclei with TDP-43 were collected (red box). (C) Gene fold changes of genes that were common significant differentially expressed linked to TDP-43 loss and the C9orf72 mutation were plotted, showing that genes that were downregulated due to the C9orf72 mutation were upregulated due to TDP-43 loss, suggesting that depletion of diseased nuclei (without TDP-43) can partially explain the transcriptomic changes observed in this dataset (Pearson’s r=-0.5708, p<0.0001). (D) Genome-wide gene fold changes that were expressed in both datasets linked to TDP-43 loss and C9orf72 mutation showed a similar pattern. Global C9orf72 associated transcriptome changes are linked to loss of function of the C9orf72 protein Current hypotheses of C9orf72 toxicity include loss of C9orf72 protein or gain of toxic RNA foci and dipeptide repeat aggregates (9). While some transcriptome changes which appear to be related to C9orf72 gain of toxic function (genes associated with proteostasis and DNA damage, dysregulation of transcripts with hnRNP 3’UTR binding sites), other changes appeared to be linked to the loss of C9orf72 protein (genes associated with vesicle membranes and endosomal trafficking). However, at a global transcriptome level, it was unclear whether the overall changes associated with the mutation reflect changes linked to a toxic gain of C9orf72 function or the loss of C9orf72 protein. C9orf72 RNA foci and dipeptide repeat protein aggregates are seen in C9orf72 repeat expansion carriers but absent in neurologically normal controls. We have also demonstrated that within C9orf72 mutation carriers, C9orf72 promoter methylation is negatively correlated with the accumulation of RNA foci and dipeptide repeat protein aggregates in human brains (43-45). Based on this, we developed a theoretical framework (Fig 4A-D) wherein statistical analysis was performed within C9orf72 expansion cases versus statistical analysis between C9orf72 expansion cases and controls. As a theoretical example, using C9orf72 itself as a gene that contributes to the loss of C9orf72 protein, we predicted that within C9orf72 expansion carriers, methylation of the C9orf72 promoter would negatively correlate with C9orf72 expression (Fig 4A). Moreover, we predicted that between-group analysis would show that C9orf72 expression is downregulated compared to controls (Fig 4B). This would be an example of a “concordant” gene which provides evidence that the altered gene expression is linked to the loss of C9orf72 protein. Conversely, using similar logic, if a gene contributes to the gain of toxic function such as the formation of toxic RNA, we would predict that within C9orf72 expansion carriers, methylation of the C9orf72 promoter would negatively correlate with toxic RNA (Fig 4C). However, between groups, this gene would be upregulated in C9orf72 expansion carriers compared to controls (Fig 4D). This would be an example of a “discordant” gene, which provides evidence that altered expression of this gene is associated with a gain of toxic function.
Fig 4: C9orf72 transcriptome reflects the loss of C9orf72 function. (A) Theoretical analysis within C9orf72 mutation carriers, we predict that C9orf72 promoter methylation would negatively correlate with C9orf72 expression. (B) Theoretical analysis between C9orf72 mutation carriers and non-diseased controls, we predict that C9orf72 expression is downregulated compared to controls. This would be an example of a “concordant” gene that suggests this gene is linked to loss of C9orf72 protein. (C) Theoretical analysis within C9orf72 mutation carriers, we predict that C9orf72 promoter methylation would negative correlate with toxic RNA. (D) Theoretical analysis between C9orf72 expansion carriers and non-diseased controls, we predict that toxic RNA expression would be upregulated. This would be an example of a “discordant” gene that suggests this gene is linked to gain of toxic RNA. (E) Within the actual data, there was a negative relationship between C9orf72 expression and methylation (Pearson r=-0.6958, p=0.0825). (F) C9orf72 expression is downregulated in C9orf72 expansion carriers compared to controls (t-test, p=0.0003) as predicted in genes associated with the loss of C9orf72 protein. (G) Within C9orf72 mutation carriers, there was a strong correlation between the expression of SYP and C9orf72 methylation (Pearson r=0.8835, p=0.0083). (H) Between C9orf72 expansion carriers and controls, there was a significant upregulation of SYP in expansion carriers (t-test, p=0.03). (I) Using significantly differentially expressed genes, the methylation correlation within C9orf72 mutation cases was plotted against gene fold change between normal C9orf72 mutation cases and controls. There were more concordant genes than discordant genes, reflecting that the transcriptome was associated with loss of C9orf72 protein. (H) Genome wide analysis of methylation correlation vs gene fold change shows the same trend. Using the actual data, a correlation analysis was performed comparing C9orf72 expression and methylation of the C9orf72 promoter within our C9orf72 expansion cohort (Pearson’s r=-0.6958; p=0.0825) (Fig 4E). Moreover, between-group analysis revealed that C9orf72 expression is downregulated in C9orf72 expansion carriers compared to controls as expected (t-test, p=0.0003) (Fig 4F). Another example of a “concordant” gene is synaptophysin (SYP), a synaptic vesicle protein. The same correlation analysis was done which showed a positive correlation within C9orf72 expansion carriers (Pearson’s r=0.8835; p=0.0083), and an increase in expression in C9orf72 expansion carriers compared to controls (t-test, p=0.03) (Fig 4G-H, supplemental table 4). Thus, we are able to show that “concordant” genes tend to reflect genes that may be related to C9orf72 protein function. This analysis was extended to all significantly differentially expressed genes where the “within C9orf72 mutation cases” methylation correlation value was plotted as a function of “between C9orf72 cases and controls” gene fold change values (Fig 4I). Across all significantly differentially expressed genes, there were 267 genes found within the concordant quadrants (blue circles) compared to only 56 genes found in the discordant quadrants (red circles) (Fig 4G). Similar results were found when this analysis was further expanded transcriptome-wide, suggesting that the global transcriptome changes linked to the C9orf72 mutation are associated with loss of C9orf72 protein (Fig 4H). Many “concordant” genes were genes involved in synaptic vesicle formation (i.e. clathrin light chain B, syntaphilin, synaptophysin) and vesicle trafficking (i.e. multivesicular body subunit 12A, RAB40B, member RAS oncogene family). Genes labeled as “discordant” include genes related to inflammatory or apoptotic processes including tec protein tyrosine kinase, tyrosine kinase, and Ring finger protein 152. Thus two different factors appear to shape this global C9orf72 transcriptome dataset: (1) the depletion of neurons with TDP-43 pathology prior to RNA-sequencing and (2) the loss of C9orf72 function. To estimate the relative contributions of these two factors, a multivariate linear regression analysis was performed wherein significant changes in gene expression associated with the C9orf72 mutation were related to (1) the changes in gene expression we previously identified due to TDP-43 pathology and (2) the loss of C9orf72 gene expression due to methylation of the C9orf72 promoter. Together, these two factors explained the majority of the variance in this dataset (R2=0.5138; βmethylation=0.838, p<2e-16; βTDP-43=-0.499, p<2e-16). Discussion Here we report that in using post-mortem neuronal nuclei without TDP-43 pathology from C9orf72 expansion carriers, we were able to identify transcriptome changes that link the C9orf72 mutation with loss of C9orf72 function. We found that the top 1% of genes that contribute most to changes in C9orf72 expression were involved in cytoplasmic vesicle trafficking. Significant gene expression changes were linked to the C9orf72 mutation and featured upregulated genes related to synaptic transmission and endosomal/lysosomal trafficking. Indeed, C9orf72 has been shown to be involved in endosomal or lysosomal trafficking and may be a Rab GTPase, and recent whole tissue transcriptome studies have linked the C9orf72 mutation to changes in vesicular transport genes (3-8). Nine of the 11 genes related to trafficking were upregulated, potentially reflecting a compensatory mechanism to counteract the loss of C9orf72 and loss of this trafficking related protein. Further analysis correlating methylation and gene expression changes confirmed that the transcriptome changes are linked to loss of C9orf72 expression. To parse the relative contributions of C9orf72 protein loss or gain of toxic RNA, a methylation correlation between methylation values and expression data was calculated. This correlation analysis indicated that the transcriptome observed in neuronal nuclei from C9orf72 expansion cases was driven in large part by the loss of C9orf72 protein as opposed to a toxic gain of function. In contrast, we observed minor splicing alterations and relatively subtle changes in terms of alterations in RNA binding protein function. Thus, the global alterations appear to be most reflective of a loss of C9orf72 function rather than toxic RNA mediated effects. However, it should be noted that C9orf72 methylation was used as a proxy for DPR and RNA foci levels. A more direct analysis of the effects of DPR or RNA foci would require specific isolation of DPR or RNA-foci containing neuronal nuclei which has not yet been possible using FAC sorting approaches. Additional future studies can also include comparisons between ALS/FTD cases with and without the C9orf72 mutation. Genes related to DNA repair either as a response to DNA damage or important to employ DNA repair were also dysregulated. Interestingly, recent papers have shown that DNA damage is activated by the C9orf72 repeat expansion in both ALS patients and experimental models (23, 25, 26, 30, 68). DNA damage has been a consistent feature among trinucleotide repeat expansion diseases and the dysregulation of DNA repair related genes is consistent with DNA damage contributing to neurodegeneration (69). Furthermore, alterations in proteostasis either by failure to fold proteins properly or failure to degrade proteins likely contribute to protein aggregation and are a common theme among all neurodegenerative diseases (70, 71). Indeed, overexpressing proteostasis factors in animal and cell ALS models prevents protein accumulation and aggregation (57, 72, 73), suggesting that altered proteostasis activity contributes to neurodegeneration. Thus, although the global transcriptome appears to be driven by the loss of C9orf72 function, embedded within the C9orf72 transcriptome are changes in gene expression, which may relate to the toxic functions of C9orf72. In comparing the data from previous transcriptome analyses from C9orf72 expansion carriers, several differences can be observed. Previous transcriptome analyses have identified pathways involved in inflammation and defense responses among dysregulated genes in the frontal cortices from C9orf72 expansion carriers (49). Within our dataset, we did not find enriched inflammatory pathways, which is likely due to the fact that only neurons were being sequenced. It is possible that because C9orf72 is expressed in microglia, C9orf72 protein function may have a larger effect on microglial populations (41). Given the evidence supporting that C9orf72 is involved inflammatory processes, further cell-specific analyses may prove beneficial to better understanding the effects of mutant C9orf72 on non-neuronal cells in driving disease pathogenesis. Previous studies have found large numbers of splicing changes in C9orf72 expansion carriers and found motifs within these splicing changes that correspond to hnRNP H binding (49). However, we did not detect such large changes and instead found milder splicing changes with motifs that do not correspond to any known RNA foci colocalization partners (data not shown). Current studies debated over the disease relevance of RNA foci as some animal models overexpressing the repeat induce RNA foci but do not show motor or cognitive defects (13-16). The lack of splicing changes in our dataset does not support the model wherein neuronal toxicity is linked to the titration of RNA binding proteins within RNA foci. Overall, our findings argue against RNA toxicity attributed to sequestration of RNA binding proteins. Other disease-related pathways, such as protein aggregation and DNA repair, were observed in this dataset, supporting a potential role for dipeptide repeat protein aggregates and nuclear DNA damage in disease, consistent with experimental studies showing that overexpression of dipeptide repeat proteins are toxic both in cells and in animal models (13-32). While the transcriptome appeared to be shaped in large part by the loss of C9orf72 function, it is unclear whether the loss of C9orf72 function contributes to disease and thus understanding whether these changes are functionally linked to disease pathogenesis requires additional experimental studies and validation. Finally, our analysis highlights the complexities associated with molecular studies of post-mortem human tissue wherein issues related to cell identity, experimental design, and admixtures of both gain and loss of function effects can be seen concurrently. Contributions and Acknowledgements These studies were supported by a grant from the Judith & Jean Pape Adams Charitable Foundation, a Clinical Scientist Development Grant from the Doris Duke Charitable Foundation, and NIH grants (T32 AG000255, R21 NS097749, R01 NS095793, P01 AG017586, and P30 AG010124). We thank Paul Hallberg from the University of Pennsylvania Flow Cytometry Core for technical assistance, and the Center for Neurodegenerative Disease at the University of Pennsylvania for their support. We are indebted to the patients, caretakers, families, and physicians, for without their meaningful contributions and dedication to medical research this work would not be possible. References 1. M. DeJesus-Hernandez et al., Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245 (Oct 20, 2011). 2. A. E. Renton et al., A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257 (Oct 20, 2011). 3. Y. Aoki et al., C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain : a journal of neurology 140, 887 (Apr 01, 2017). 4. M. A. Farg et al., C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23, 3579 (Jul 01, 2014). 5. D. Zhang, L. M. Iyer, F. He, L. Aravind, Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet 3, 283 (2012). 6. D. Tang et al., Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci U S A 117, 9876 (May 5, 2020). 7. C. P. Cali et al., C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathologica 138, 795 (Nov, 2019). 8. D. W. Dickson et al., Extensive transcriptomic study emphasizes importance of vesicular transport in C9orf72 expansion carriers. Acta Neuropathol Commun 7, 150 (Oct 8, 2019). 9. S. C. Ling, M. Polymenidou, D. W. Cleveland, Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416 (Aug 7, 2013). 10. J. Cooper-Knock et al., Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain : a journal of neurology 137, 2040 (Jul, 2014). 11. Y. B. Lee et al., Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5, 1178 (Dec 12, 2013). 12. E. G. Conlon et al., The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5, (Sep 13, 2016). 13. J. G. O'Rourke et al., C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 88, 892 (Dec 02, 2015). 14. O. M. Peters et al., Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 88, 902 (Dec 02, 2015). 15. A. Jain, R. D. Vale, RNA phase transitions in repeat expansion disorders. Nature 546, 243 (Jun 08, 2017). 16. H. Tran et al., Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 87, 1207 (Sep 23, 2015). 17. B. D. Freibaum et al., GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129 (Sep 03, 2015). 18. A. Jovicic et al., Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18, 1226 (Sep, 2015). 19. S. May et al., C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128, 485 (Oct, 2014). 20. Z. Tao et al., Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet 24, 2426 (May 01, 2015). 21. X. Wen et al., Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213 (Dec 17, 2014). 22. L. D. Goodman et al., Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nature neuroscience 22, 863 (Jun, 2019). 23. Y. J. Zhang et al., Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363, (Feb 15, 2019). 24. Y. J. Zhang et al., Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24, 1136 (Aug, 2018). 25. Y. Nihei et al., Poly-glycine-alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathologica 139, 99 (Jan, 2020). 26. R. Lopez-Gonzalez et al., Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc Natl Acad Sci U S A 116, 9628 (May 7, 2019). 27. T. G. Moens et al., C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathologica 137, 487 (Mar, 2019). 28. A. Jovicic et al., Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nature neuroscience 18, 1226 (Sep, 2015). 29. K. Zhang et al., The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56 (Sep 3, 2015). 30. R. Lopez-Gonzalez et al., Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 92, 383 (Oct 19, 2016). 31. S. Mizielinska et al., C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192 (Sep 05, 2014). 32. Y. Ohki et al., Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol Neurodegener 12, 6 (Jan 14, 2017). 33. P. E. Ash et al., Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639 (Feb 20, 2013). 34. M. DeJesus-Hernandez et al., In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol 134, 255 (Aug, 2017). 35. I. R. Mackenzie et al., Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 130, 845 (Dec, 2015). 36. T. G. Moens et al., Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathologica 135, 445 (Mar, 2018). 37. A. J. Waite et al., Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiology of aging 35, 1779 e5 (Jul, 2014). 38. P. Frick et al., Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun 6, 72 (Aug 3, 2018). 39. A. Burberry et al., Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8, 347ra93 (Jul 13, 2016). 40. M. Koppers et al., C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals of neurology 78, 426 (Sep, 2015). 41. J. G. O'Rourke et al., C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324 (Mar 18, 2016). 42. Q. Zhu et al., Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nature neuroscience 23, 615 (May, 2020). 43. E. Y. Liu et al., C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 128, 525 (Oct, 2014). 44. C. T. McMillan et al., C9orf72 promoter hypermethylation is neuroprotective: Neuroimaging and neuropathologic evidence. Neurology 84, 1622 (Apr 21, 2015). 45. J. Russ et al., Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol 129, 39 (Jan, 2015). 46. K. J. Karczewski et al., The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434 (May, 2020). 47. R. H. Batra, K.; Vu, A.;Rabin, S.J.; Baughn, M.W.; Libby, R.T.; Hoon, S.; Ravits, J.; Yeo, G.W., Gene Expression Signatures of Sporadic ALS Motor Neuron Populations. bioRxiv, (2016). 48. J. Cooper-Knock et al., C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS One 10, e0127376 (2015). 49. M. Prudencio et al., Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci 18, 1175 (Aug, 2015). 50. F. Krach et al., Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathologica 136, 405 (Sep, 2018). 51. S. Curran, J. A. McKay, H. L. McLeod, G. I. Murray, Laser capture microscopy. Mol Pathol 53, 64 (Apr, 2000). 52. E. Y. Liu et al., Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell reports 27, 1409 (Apr 30, 2019). 53. J. B. Toledo et al., A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 10, 477 (Jul, 2014). 54. J. R. Tollervey et al., Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452 (Apr, 2011). 55. S. C. Huelga et al., Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1, 167 (Feb 23, 2012). 56. J. Wang, D. Duncan, Z. Shi, B. Zhang, WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic acids research 41, W77 (Jul, 2013). 57. H. J. Chen et al., The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain : a journal of neurology 139, 1417 (May, 2016). 58. D. Chhangani et al., Mahogunin ring finger 1 confers cytoprotection against mutant SOD1 aggresomes and is defective in an ALS mouse model. Neurobiol Dis 86, 16 (Feb, 2016). 59. A. M. Ali et al., FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood 119, 3285 (Apr 05, 2012). 60. Y. Birger et al., Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. The EMBO journal 22, 1665 (Apr 01, 2003). 61. Y. Nomura, N. Adachi, H. Koyama, Human Mus81 and FANCB independently contribute to repair of DNA damage during replication. Genes Cells 12, 1111 (Oct, 2007). 62. B. Wang et al., Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194 (May 25, 2007). 63. L. Castellini et al., KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic acids research 45, 3674 (Apr 20, 2017). 64. L. Miccoli et al., Selective interactions of human kin17 and RPA proteins with chromatin and the nuclear matrix in a DNA damage- and cell cycle-regulated manner. Nucleic acids research 31, 4162 (Jul 15, 2003). 65. L. C. Young, D. W. McDonald, M. J. Hendzel, Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. The Journal of biological chemistry 288, 21376 (Jul 19, 2013). 66. B. Chen, J. Yun, M. S. Kim, J. T. Mendell, Y. Xie, PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol 15, R18 (Jan 22, 2014). 67. E. B. Lee et al., Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathologica 134, 65 (Jul, 2017). 68. M. A. Farg, A. Konopka, K. Ying Soo, D. Ito, J. D. Atkin, The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in Amyotrophic Lateral Sclerosis. Hum Mol Genet, (May 08, 2017). 69. A. Lopez Castel, J. D. Cleary, C. E. Pearson, Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11, 165 (Mar, 2010). 70. P. M. Douglas, A. Dillin, Protein homeostasis and aging in neurodegeneration. J Cell Biol 190, 719 (Sep 06, 2010). 71. O. D. King, A. D. Gitler, J. Shorter, The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 1462, 61 (Jun 26, 2012). 72. M. E. Jackrel et al., Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156, 170 (Jan 16, 2014). 73. P. Y. Lin et al., Heat shock factor 1 over-expression protects against exposure of hydrophobic residues on mutant SOD1 and early mortality in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener 8, 43 (Nov 21, 2013).
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |