|
|
|
Free Neuropathology 1:8 (2020) |
|
Review |
|
Top ten discoveries of the year: Neurooncology |
|
Pieter Wesseling1,2 |
|
1 Department of Pathology, Amsterdam University Medical Centers, location VUmc, Brain Tumor Center Amsterdam, De Boelelaan 1117, 1081HV Amsterdam, The Netherlands |
|
Corresponding author: |
|
Submitted: 30 January 2020 Accepted: 23 February 2020 Published: 26 February 2020 |
|
Keywords: Brain tumor, Neuropathology, Molecular diagnostics, Glioma, Medulloblastoma, Methylome analysis |
|
Abstract This article briefly discusses 10 topics that were selected by the author as top 10 discoveries published in 2019 in the broader field of neuro-oncological pathology (so including neurosciences as well as clinical neuro-oncology but with implications for neuro-oncological pathology). Some topics concern new information on immunohistochemical and molecular markers that enable improved diagnosis of particular tumors of the central nervous system (CNS) and information on a refined classification of medulloblastomas. Subsequently, several papers are discussed that further elucidate some pathobiological aspects of especially medulloblastomas (histogenesis, molecular evolution) and diffuse gliomas (mechanisms involved in CNS infiltration, role of cancer stem(-like) cells, longitudinal molecular evolution). The remaining topics concern progress made in vaccination therapy for glioblastomas and in using cerebrospinal fluid for liquid biopsy diagnosis of gliomas. Clearly, substantial, and sometimes even amazing progress has been made in increasing our understanding in several areas of neuro-oncological pathology. At the same time, almost every finding raises new questions, and translation of new insights in improving the outcome for patients suffering from CNS tumors remains a huge challenge. Introduction Looking back from time to time is good. The request of the editor-in-chief of this journal to contribute a review on ‘top 10 discoveries’ in neuro-oncology published in 2019 was the reason for the author to look back in a somewhat more structured way in order to select which topics could/should qualify for such a label. From the start it was clear that there are multiple ways to shape such a selection process. To cut a long story short, keeping the readership of Free Neuropathology in mind and with some help of the editor-in-chief of this journal, the choice was made to select topics based on original papers published in 2019 from a broad range of journals, important criteria being that indeed relevant progress was made and that in one way or another the findings can be expected to have substantial implications for neuro-oncological pathology. Furthermore, it was decided to not just focus on articles in the high(est)-impact journals and to aim for a somewhat broader blend of topics.
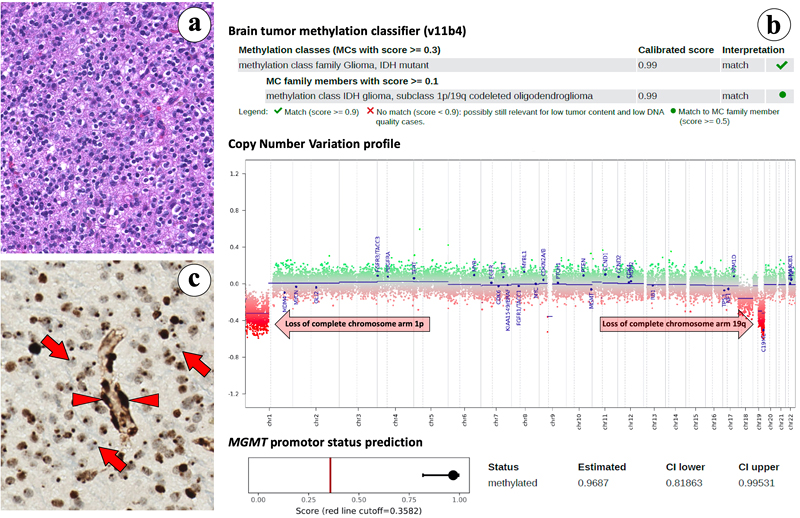
Some topics presented in this review were selected based on just one article, while the information of two or three papers was combined for the discussion of other topics. The 10 topics that emerged from this (quite subjective!) selection process are: Of note, the ranking in this list is not based on importance of the findings, but rather on an attempt to create a flow in this review (from information that may quite readily be implemented in daily diagnostic practice via more hard-core neurobiology/neuroscience findings to some future perspectives). Also, there is a bias in the selection of the topics in this list towards diffuse gliomas and embryonal tumors/medulloblastomas. Obviously, other colleagues might have presented a quite different list of ‘top 10 discoveries’, also because many more very good and interesting papers with implications for neuro-oncological pathology were published in 2019. For example, the study of Paramasivam et al [18], providing novel information on mutational patterns and regulatory networks of subgroups of meningiomas would be a good alternative for a topic. Having said that, it is hoped that this review not only provides easily digestible information on the top 10 discoveries in neuro-oncological pathology as selected by this author, but will also be used as a stepping stone to appreciate the much more detailed information in the original papers that were used as building blocks for this review. Topic 1. Lack of H3K27me3 staining is a promising surrogate marker for oligodendrogliomas [1] Unequivocal histological recognition of (anaplastic) oligodrogliomas, IDH-mutant & 1p/19q-codeleted (‘canonical oligodendrogliomas’) has been hindered by the lack of specific immunohistochemical markers for these tumors [19]. In a study analyzing an epigenetically well-defined cohort of diffuse gliomas in adults (IDH-mutant & 1p/19q-codeleted oligodendrogliomas, n = 26; IDH-mutant astrocytic tumors, n = 34; IDH-wildtype glioblastomas, n = 101), Filipski et al report that 25 out of the 26 oligodendrogliomas showed lack of nuclear H3K27me3 staining, in the remaining case H3K27me3 staining was interpreted as non-conclusive [1]. Interestingly, in an otherwise ‘nucleonegative’ oligodendroglioma of a female patient only dot-like nuclear staining was seen. This phenomenon has previously been described to represent the inactivated X chromosome, with H3K27me3 functioning as a transcriptional silencing mechanism via chromatin remodeling [20] (Fig. 1). The vast majority of IDH-mutant and IDH-wildtype astrocytic tumors showed clear nuclear H3K27me3 staining. Based on these findings in combination with the results of immunohistochemistry for ATRX, IDH1R132H and H3K27M, Filipski et al report that in their cohort of H3K27-wildtype diffuse gliomas the tumors showing lack of nuclear H3K27me3 staining, retention or non-conclusive nuclear ATRX staining, and IDH1R132H mutation are oligodendroglioma, IDH-mutant & 1p/19q codeleted with a predicted probability of 0.9678. Ideally, in case of H3K27me3- and H3K27M-nucleonegative diffuse gliomas that in addition show retained or non-conclusive ATRX staining but no IDH1R132H-mutant protein staining, sequencing analysis is performed to demonstrate presence/absence of an IDH1 or IDH2 mutation. Obviously, the results of immunohistochemistry should be interpreted with caution in especially biopsies that are small and/or only show low tumor cell percentage. The use of an antibody panel including H3K27me3, H3K27M mutant protein, ATRX, and IDH1R132H may thus greatly facilitate recognition (anaplastic) oligodendrogliomas, IDH-mutant & 1p/19q-codeleted, especially so in a situation where molecular testing is not readily available/possible. The pathobiology underlying the global lack of H3K27me3 in tumor cell nuclei and 1p/19q codeletion needs further elucidation. Of note, while in diffuse gliomas in adults lack of H3K27me3 staining may thus indicate canonical oligodendroglioma with a relatively favorable prognosis, in several other tumors of the (central) nervous system (e.g. posterior fossa ependymomas, malignant peripheral nerve sheath tumors, meningiomas) such a lack of nuclear staining is associated with worse prognosis [21-23].
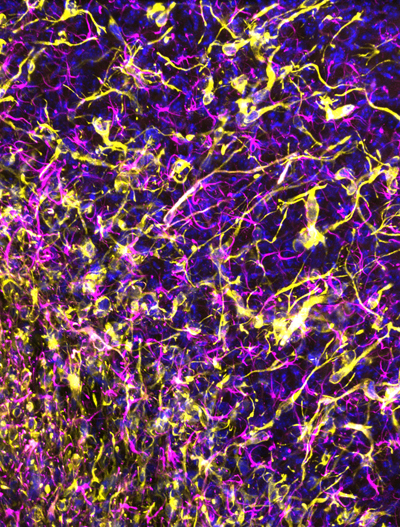
Fig. 1. Loss of nuclear H3K27me3 immunohistochemical staining; a surrogate marker for oligodendroglioma, IDH-mutant and 1p/19q-codeleted. Topic 2. Improved molecular diagnosis of rare CNS tumor types [2, 3] Only recently, genome-wide DNA methylation analysis has been introduced as a very helpful tool for improved diagnosis of CNS tumor types and subtypes [24-29]. In 2019, two papers were published by the ‘Heidelberg team’ + coauthors from other centers/countries demonstrating the power of methylome analysis. Sievers et al [2] report the results of in-depth analysis of 30 tumors initially identified through methylome analysis as a separate group in a Heidelberg cohort of > 25,000 tumors. Most tumors in this group were histologically diagnosed as rosette-forming glioneuronal tumor (RGNT), and all revealed FGFR1 hotspot mutations, with in about two-thirds of the cases co-occurrence of a PIK3CA mutation and in one third of the tumors an additional loss-of-function mutation in NF1. In contrast to most low-grade gliomas, RGNTs displayed co-occurrence of two or even all three of these mutations. These highly recurrent combined genetic alterations affecting both MAPK and PI3K signaling pathways may offer potential therapeutic targets for RGNTs [2]. In a study of Hou et al [3], 17 of the 28 tumors initially histologically diagnosed as papillary glioneuronal tumor (PGNT) showed methylation profiles typical for other tumor entities (mostly dysembryoplastic neuroepithelial tumor and pilocytic astrocytoma). The remaining 11 cases exhibited a unique profile and were considered as a distinct PGNT methylation class. Three additional tumors in the Heidelberg cohort clustered with this methylation class as well but were originally not diagnosed as PGNT. In all 12 cases of this methylation class of which material was available for further analysis, fusions involving PRKCA were identified (SLC44A1–PRKCA, n = 11; NOTCH1-PRKCA, n = 1), whereas such fusions were not found in the tumors belonging to other methylation classes. Both the study of Sievers et al and of Hou et al thus provide information on molecular characteristics that can be used for improved recognition of such rare CNS tumor types and again illustrate the power of methylome analysis in this context. Topic 3. Refined molecular classification of non-WNT/non-SHH medulloblastomas [4] The 2016 WHO Classification of CNS tumors recognizes four molecular variants of medulloblastoma: WNT-activated, SHH-activated and TP53-mutant, SHH-activated and TP53-wildtype, and non-WNT/non-SHH. The non-WNT/non-SHH subgroup, accounting for about 65% of all medulloblastomas, encompasses the Group 3 and Group 4 molecular variants of medulloblastoma. However, these groups have heterogeneous clinical characteristics and survival outcomes, and their biology remained less clear. Studies published in 2017 suggested the existence of 4, 6 or 8 subgroups within the overarching non-WNT/non-SHH group [30-32]. In an attempt to provide clarity and a basis for further biological studies, Sharma et al analyzed the number and nature of subtypes that could be identified in a cohort of 1501 genomically characterized non-WNT/non-SHH medulloblastomas [4]. In this study, rather than advocating one single analytical approach or method, multiple class-definition approaches were used and equal weight was given to each analytical technique. In a lower complexity analysis Group 3 and Group 4 were identified again, but more complex analysis strongly supported the existence of eight robust Group 3/Group 4 subtypes (I-VIII). These subtypes could generally be recognized based on their DNA-methylation profiles and showed differences in enrichment for specific driver gene alterations, cytogenetic events, and ages of incidence with mostly unimodal age distribution. Cytogenetic signatures, chromosomal copy-number aberrations and information on one gene or a set of genes in isolation were found to be insufficient for recognition of subtypes I-VIII. The relevance of these subtypes is supported by differences in survival, with e.g. subtype IV being associated with low-risk clinical behavior, and with relatively frequent late relapses in patients with a subtype VIII medulloblastoma. These findings can be expected to improve risk stratification, therapy and thereby the outcome for patients with non-WNT/non-SHH medulloblastomas. Of note, although the bimodal age distribution that was found for subtypes V and VII may indicate that some further refinement of the classification is still possible, the authors report that they did not find strong evidence for more than eight molecular subgroups in the non-WNT/non-SHH category. Subtyping of non-WNT/non-SHH medulloblastomas into subtype I-VIII is now available for the community by performing methylome analysis as described by Capper et al [24, 25] and using an extension of the Heidelberg brain tumor classifier (https://www.molecularneuropathology.org/mnp/classifier/7). Topic 4. Histogenesis of posterior fossa tumors in children [5-7] For quite some time it is clear that different subtypes of medulloblastomas have different developmental origin [33]. In fact, cerebellar tumors may be the result of a disorder of early cerebellar development. In an attempt to further elucidate cerebellar development at the single cell level, Vladiou et al performed large-scale single-cell RNA sequencing (scRNA-seq) of more than 60,000 cells from the developing mouse cerebellum [5]. These analyses allowed them to reconstruct the cellular hierarchy in development at various points in time, with many of the normal mouse cerebellar cell populations only being present for a restricted time period during the fetal or very early postnatal period. Subsequently, this transcriptome information was compared to that of pediatric cerebellar tumors: medulloblastomas, posterior fossa (PF) ependymomas, and pilocytic astrocytoma. The authors find that (different molecular subgroups of) these tumors indeed mirror the transcription of cells from distinct, temporally restricted cerebellar lineages: SHH medulloblastomas, Group 3 and Group 4 medulloblastomas were found to transcriptionally resemble respectively the granule cell hierarchy, Nestin+ stem cells, and unipolar brush cells, while PF type A/PF type B (PFA/PFB) ependymomas and cerebellar pilocytic astrocytomas resembled prenatal gliogenic progenitor cells. These findings indicate that each of these cerebellar tumor types arises from a particular cell type. However, across the medulloblastoma subgroups (SHH, Group 3, Group 4), as well as PFA ependymoma and pilocytic astrocytomas, the scRNAseq data also demonstrated high levels of single-cell heterogeneity, with evidence of multiple lineages of differentiation and tumor cells matching different time points in the differentiation hierarchy [5]. In another study, Hovestadt et al performed single-cell transcriptomics to investigate intra- and intertumoral heterogeneity in 25 medulloblastomas spanning all molecular subgroups. They find that WNT, SHH and Group 3 tumors comprise subgroup-specific undifferentiated and differentiated neuronal-like populations of malignant cells, whereas Group 4 tumors consist of differentiated neuronal-like neoplastic cells. The tumor cells in SHH medulloblastomas closely resembled granule neurons of varying differentiation states that correlated with patient age. Using cross-species transcriptomic analysis, it appeared that distinct glutamatergic populations are the putative cells-of-origin for SHH and Group 4 medulloblastomas. Furthermore, the tumor cells of Group 3 and Group 4 medulloblastomas exhibited characteristics ranging from primitive progenitor-like to more mature neuronal-like cells, with the relative proportions of these cells distinguishing these subgroups [6]. In a third study, based on single-cell transcriptome analysis of >65,000 cells of the embryonal pons and forebrain, Jessa et al derived signatures for 191 distinct cell populations of these regions [7]. Bulk transcriptome analysis of WNT-activated medulloblastomas revealed a match with the mossy fiber neuronal lineage of the rhombic lip. Embryonal tumors with multilayered rosettes (ETMRs) appeared to be derived from a neuronal lineage as well, but especially the TYR and MYC subgroups of atypical teratoid/rhabdoid tumor (AT/RT) seemed to originate from non-neuroectodermal cells. All in all, these studies provide strong evidence that pediatric CNS tumors are the result of a disorder of early development, although the possibility that they arise from more mature cells undergoing de-differentiation at later time points or from trans-differentiation of other cell lineages cannot yet be completely ruled out. Topic 5. Longitudinal molecular analysis of diffuse gliomas [8] Genomic characterization efforts such as The Cancer Genome Atlas (TCGA) have greatly increased our understanding of glioma biology [34-37]. Based on these findings three major, clinically relevant subgroups of diffuse gliomas in adults were introduced in the WHO 2016 classification of CNS tumors: (1) IDH-mutant and chromosome 1p/19q codeleted (IDH-mutant-codel); (2) IDH-mutant without codeletion of chromosome 1p/19q (IDH-mutant-noncodel); and (3) IDH-wildtype. So far, however, such studies were generally limited to analysis of tumor tissue as obtained by first operation, and how the genetic landscape of these gliomas evolves over time and in response to therapy remained largely unknown. In December 2019, the Glioma Longitudinal AnalySiS (GLASS) Consortium published the results of analysis of 222 diffuse glioma patients (25 IDH-mutant-codel, 63 IDH-mutant-noncodel, and 134 IDH-wildtype) with high-quality data on mutations and chromosomal copy numbers in samples of at least two time points [8]. The study revealed that the driver genes detected in the initial sample were retained in the recurrent tumor, and there was little evidence of recurrence-specific gene alterations. Also, current standard of care therapies (chemotherapy, irradiation) generally did not seem to coerce the tumors down predictable paths. IDH-mutant-noncodels were found to be most sensitive to developing a hypermutator phenotype after therapy with alkylating agents, a phenomenon that has been reported before [38, 39]. Importantly, no differences were found in overall survival between hypermutators and non-hypermutators independent of age, subtype and MGMT promoter methylation status. In line with a recent study demonstrating that homozygous CDKN2A loss is a marker for high-grade malignancy in IDH-mutant-noncodels [40], in the GLASS study recurrent IDH-mutant-noncodels were enriched for homozygous CDKN2A deletions and this was associated with shorter survival compared to patients without these alterations. No differences in the levels of immunoediting were found between initial and recurrent gliomas. The GLASS study thus indicates that the strongest selective pressure in these three major subgroups of diffuse gliomas occurs during early glioma development and that current therapies shape this evolution in a largely stochastic manner. Hopefully, such information on how diffuse gliomas evolve over time and in response to therapy will help to design more efficacious therapeutic strategies for these tumors. Topic 6. NOTCH1-SOX2 signaling controls glioma cell invasion in white matter tracts [9] One of the main reasons that patients with diffuse gliomas so far cannot be cured is the diffuse infiltrative growth of the tumor cells along white matter tracts. One would hope that unraveling the mechanisms underlying this phenomenon may help to identify novel therapeutic targets. In an analysis of human glioma tissue samples, Wang et al found that at the invasive front, CD133+ ‘glioma stem cells’ (GSCs) were preferentially located along white matter tracts [41]. Also, these tracts showed significant swelling and seemed to have discontinuous myelin, indicating that some GSCs in the glioma-brain interface are preferentially located adjacent to unmyelinated white matter tracts. Based on their findings the authors postulate that glioma invasion along these tracts occurs along axons inside myelin sheaths rather than along the outer surface of such sheaths, and that glioma-associated edema may play a role in local destruction of white matter fibers and thereby pave the way for direct interaction of glioma cells and axons. Additionally, based on their findings in preclinical experiments that nearly all CD133+ GSCs were also Notch1+ and that nerve fibers at the invasive frontier all expressed Jagged1, Wang et al postulate that the interaction between Jagged1 and Notch1 may be an important determinant for the distribution of GSCs. Indeed, axonally expressed Jagged1 was found to activate the Notch signaling pathway in GSCs and to subsequently promote the transcription of SOX2 via SOX9. Conversely, SOX2 upregulation decreased the methylation of the NOTCH1 promoter to reinforce the high expression of NOTCH1 in GSCs and facilitate their white matter-tract tropism, while inhibition of Notch signaling was found to attenuate this tropism. The findings of Wang et al indicate that the NOTCH1-SOX2 positive-feedback loop is an important determinant of GSC invasion along white matter tracts, and that molecules in this loop may be exploited as therapeutic targets. However, the authors acknowledge that the mechanisms underlying glioma cell invasion along white matter tracts are probably more complex, and may also involve mechanisms as discussed in the next topic (Topic 7). Topic 7. ‘Synaptic cooption’ in glial and metastatic CNS tumors [10-12] After reporting earlier that neuroligin-3 secreted by active neurons promotes glioma growth [42, 43], in a recent study published in Nature, Venkatesh et al found that electrochemical communication exists between neurons and glioma cells through bona fide AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor-dependent synapses between these cells [10]. Also, neuronal activity was found to evoke non-synaptic activity-dependent potassium currents that were amplified by gap junction-mediated tumor interconnections and resulting in an electrically coupled network. Depolarization of glioma membranes was found to promote proliferation, whereas blocking electrochemical signaling inhibited the growth of glioma xenografts in mice. Intraoperative electrocorticography in glioma patients revealed increased cortical excitability in the glioma-infiltrated brain areas. Based on these findings the authors suggest that glioma growth is promoted by synaptic and electrical integration into neural circuits, with glioma-induced increase in neuronal excitability and activity-regulated glioma growth as elements of a positive feedback loop. In the same issue of Nature, Venkaratamani et al also report that functional synapses between neurons and glioma cells exist [11]. Previously, these authors already demonstrated that many tumor cells in astrocytic tumors including glioblastomas extend ultra-long membrane protrusions (microtubes) that are used for brain invasion and proliferation, and that interconnect over long distances [44] (Fig. 2). In their recent study, Venkaratamani et al show the presence of ‘neurogliomal synapses’ in human glioma samples and in different disease models. Such synapses were found to be located on tumor microtubes and produce postsynaptic currents that are mediated by glutamate receptors of the AMPA subtype. In their experiments, neuronal activity led to synchronized calcium transients in tumor-microtube-connected glioma networks. Furthermore, perturbation of AMPA receptors and use of an AMPA receptor antagonist reduced calcium-related invasiveness of tumor cells and of glioma growth. Intriguingly, in a third study published in that same issue of Nature, Zeng et al demonstrate the existence of ‘pseudo-tripartite synapses’ between breast cancer cells and glutamatergic neurons and show that such cancer cells in the brain can co-opt a neuronal signaling pathway involving activation by glutamate ligands of N-methyl-D-aspartate receptors (NMDARs) [12]. These three studies thus suggest very peculiar brain tumor-microenvironment interactions in the form of direct, biologically relevant synaptic communication between neurons and tumor cells with potential therapeutic implications.
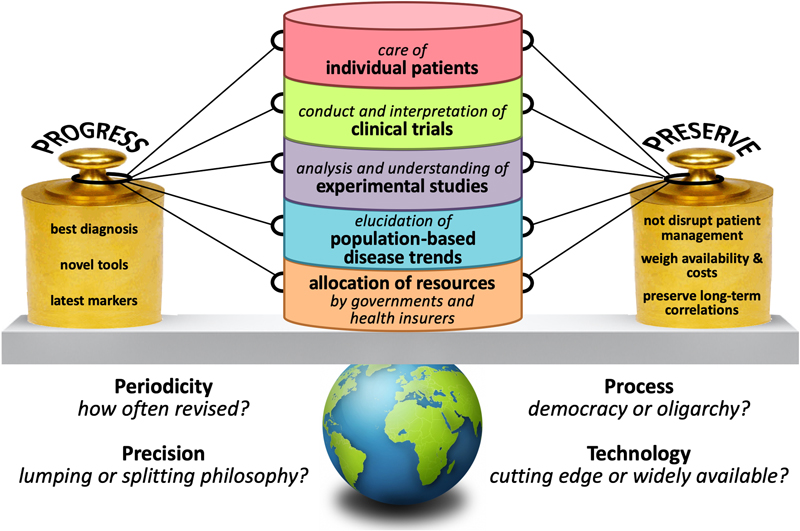
Fig. 2. Human glioma cells forming a mycelium-like network in mouse brain. Topic 8. Cancer stem cells in glioblastomas; do they exist? [13] It is been proposed that in glioblastomas cancer stem cells (CSCs) or stem-like cells reside at the top of a hierarchical organization, are able to (re)create intra-tumoral heterogeneity by generating more differentiated offspring, and are relatively resistant to therapy and thus contribute significantly to tumor recurrence [45]. Expression of particular cell membrane antigens (CD133, CD15/SSEA, CD44, and/or A2B5) has often been used for identification of the CSC subset in glioblastomas. However, it is increasingly clear that no single marker allows for unequivocal identification of a CSC population in glioblastomas. Moreover, there is still controversy if a bona fide CSC population exists at all (and if so, if it concerns a quiescent or proliferative subpopulation). In this context, Dirkse et al addressed the question whether glioblastoma cells expressing CSC-associated cell membrane markers are indeed a defined entity at the apex of a hierarchical organization. They found that, like in patient biopsies, CSC markers were heterogeneously expressed in patient-derived glioblastoma xenografts and stem-like cell cultures. More precisely, all FACS-sorted subpopulations of tumor cells of glioblastomas were able to self-renew over multiple passages without significant differences between each other. This suggests that phenotypically heterogeneous glioblastoma cells can adapt to a variety of environmental changes and acquire similar stem cell properties in vitro. Dirkse et al also found that accelerated reconstitution of heterogeneity provided a growth advantage in vivo, suggesting that tumorigenic potential is linked to intrinsic plasticity rather than CSC multipotency. The study of Dirkse et al thus provides strong evidence that CSCs do not constitute a defined cellular entity, but rather a cellular state adapting to microenvironmental cues. The capacity of any given tumor cell to reconstitute heterogeneity in glioblastomas cautions against therapies targeting only a small subpopulation of tumor cells with stem cell(-like) characteristics. Meanwhile, inherent cancer cell plasticity emerges as a novel relevant target for treatment [13]. Topic 9. Personalized (neo)antigen vaccination therapy for glioblastoma [14, 15] In patients with e.g. melanomas, high tumor mutational load (TML) is associated with increased frequency of neoantigens and improved response to checkpoint inhibition. Administration of personalized neoantigen vaccines has been shown to successfully recruit T cells to the tumor and can lead to tumor regression. Unfortunately, also after the introduction of immunotherapies, so far the very poor survival rates for patients with glioblastoma have not improved much. Of note, glioblastomas often have a relatively low mutational burden and are considered as immunologically ‘cold’. Recent studies suggest that for patients with glioblastoma, a personalized molecular approach is needed to improve the benefit of treatment with programmed cell death protein 1 (PD-1) inhibitors nivolumab or pembrolizumab, and that the neoadjuvant administration of PD-1 blockade may represent a more efficacious approach for these tumors [46, 47]. Two recent studies published in Nature explored the feasibility of vaccination therapy using tumor (neo)antigens for patients with glioblastoma. Keskin et al performed a phase I/Ib multi-epitope, personalized neoantigen vaccination study [14]. Patients who did not receive dexamethasone generated circulating polyfunctional neoantigen-specific CD4+ and CD8+ T cell responses that were enriched in a memory phenotype. After vaccination, neoantigen-specific T cells were found to migrate from peripheral blood into the glioblastoma, suggesting that they may favorably alter the immune milieu of the tumor [14]. Hilf et al studied vaccination using both unmutated tumor antigens and neoepitopes for more effective immunotherapy of glioblastomas, including those with a low mutational load [15]. Highly individualized vaccinations with both types of tumor antigens were integrated into standard care for patients with newly diagnosed glioblastoma. Fifteen patients were first treated with a vaccine with a previously constructed library of non-mutated antigens that are over-represented in glioblastomas (APVAC1), followed by a second vaccine (APVAC2) targeted against mutated neoantigens or non-mutated antigens that were not present in APVAC1. The vaccines were personalized by analysis of mutations and of the transcriptomes and immunopeptidomes of the individual tumors. Both vaccines showed favorable safety and elicited T cell responses against the proteins in the vaccine, with APVAC1 inducing a sustained CD8+ T cell response, and APVAC2 both CD4+ and CD8+ T cell responses [15]. Topic 10. Liquid biopsy diagnosis of gliomas using CSF [16, 17] Cancer cells release nucleic acids, vesicles, proteins, and other components into the blood stream and other body fluids. Already for quite some time, circulating tumor DNA (ctDNA) in blood is considered as an easily accessible source of potentially very useful diagnostic, prognostic and/or predictive information that can be used to improve the management of cancer patients [48]. So far, however, liquid biopsy diagnosis for detection and monitoring of patients with gliomas has not yet entered the clinic. Part of the problem may be that, compared to other cancers, gliomas release relatively limited amounts of ctDNA into the bloodstream [49]. Sequencing of ctDNA from the cerebrospinal fluid (CSF) may provide an alternative way to diagnose glial and other CNS tumors. Indeed, in 42 out of 85 adult patients with diffuse glioma (49.4%), Miller et al detected ctDNA in CSF that was obtained by lumbar puncture [16]. Presence of ctDNA was associated with disease burden and adverse outcome. The glioma genomes detected in CSF closely resembled those in matched tumor samples. Alterations occurring early in gliomagenesis (IDH1/IDH2 mutation, 1p/19q codeletion) were shared in all matched ctDNA-positive CSF-tumor pairs, whereas growth factor receptor signaling pathways showed considerable evolution. Most patients with ctDNA-positive CSF did not have detectable malignant cells in the CSF, and no significant association was found between ctDNA-positive CSF and glioma grade, disease duration or prior therapy. Of note, in this study of Miller et al the CSF samples were derived from patients relatively late in their disease course (median disease duration before CSF collection for patients with IDH-wildtype glioblastomas about a year, and for patients with IDH-mutant lower grade gliomas over 5 years), and all patients already had treatment for their glial tumor. In contrast, Pan et al explored the potential of ctDNA analysis in CSF obtained prior to surgical manipulation in a cohort of 57 patients with brainstem glioma [17]. Over 90% of these CSF samples were obtained intraoperatively. Pan et al report that alterations were identified in the CSF ctDNA in 36/37 cases (97.3%) in which the primary tumors harbored at least one mutation, while in 31/37 of cases (83%) all primary tumor alterations were detected in the CSF. In these patients, mutation detection using plasma ctDNA was found to be much less sensitive than sequencing the CSF ctDNA. These studies thus suggest that indeed CSF may be a much more promising biosource for liquid biopsy diagnostics of gliomas than blood. Combination of such an approach with analyses going beyond DNA sequence information (e.g. analysis of epigenetic and immune signatures in cell free DNA) may further boost the exploitation of liquid biopsy diagnostics in patients with (glial) CNS tumors [49]. Discussion The papers discussed in this review as top 10 discoveries illustrate the substantial and sometimes even amazing progress that has been made in increasing our understanding of particular topics in neuro-oncological pathology. Hopefully, this review not only provides easily digestible information on the topics selected by the author, but also works as a stepping stone to read and appreciate the original papers that were used as building blocks for the present manuscript. Obviously, most of the findings presented here immediately elicit next questions. For example, are the findings presented by Filipski et al already ‘mature’ enough to be used as surrogate markers for the diagnosis of (anaplastic) oligodendroglioma, IDH-mutant and 1p/19q-codeleted in clinical practice? And while Wang et al report that the NOTCH1-SOX2 positive-feedback loop is an important (and possibly targetable) determinant of invasion of glioma cells along white matter tracts, the authors acknowledge that other mechanisms as discussed in Topic 7 may be involved as well. But then again, is synapse formation between neuronal and glioma cell processes especially relevant for the invasive front? Or also for proliferation of tumor cells in the highly cellular center of glioblastomas where neurites can be expected to be (much) more scarce? What about the presence of ‘pseudo-tripartite synapses’ in the ‘bulky’ areas of metastatic breast cancer in the CNS? And (see Topic 8): Do bona fide glioma stem cells exist? Nowadays, a rapidly increasing number of diagnostic and prognostic molecular markers can be used for an improved, ‘histo-molecular’ diagnosis of CNS tumors [50]. Multiple assays have been developed for this purpose, ranging from single gene tests to high-throughput, integrated techniques enabling detection of multiple genetic aberrations in a single workflow [51]. Immunohistochemistry is a helpful, relatively inexpensive alternative for further molecular characterization of particular CNS tumors. Within a few years after its introduction, genome-wide methylation profiling has already had a revolutionary impact on CNS tumor classification [24, 25, 52]. The papers discussed in Topic 2 (‘Improved molecular diagnosis of rare CNS tumor types’) provide examples of the strength of this platform. However, such methylome analysis is relatively expensive. Obviously, a test providing clinically very valuable information at low costs will much more easily be accepted as a routine diagnostic tool compared to an expensive test with limited clinical benefits. Unfortunately, a clear framework for assessment of cost-effectiveness of molecular testing of CNS (and other) tumors is lacking [53]. It is important to realize though that molecular diagnostics of (CNS) tumors is relatively inexpensive compared to e.g. neuro-imaging and chemotherapy, and that a diagnosis that is ‘on target’ will not only have substantial clinical benefit for the patients but may also help to avoid unnecessary health care costs [52, 54]. Meanwhile, since the publication of the WHO Classification of CNS Tumours in 2016, the consortium to Improve Molecular and Practical Approaches into CNS tumor Taxonomy (cIMPACT-NOW) has already published several updates with suggestions on how exactly particular molecular markers can be used for improved diagnosis in clinical practice (see for summary of round 1 updates [55]). The findings described in Topic 3 (‘Refined molecular classification of non-WNT/non-SHH medulloblastomas‘) and Topic 4 (‘Histogenesis of posterior fossa tumors in children’) may soon be incorporated into a more refined next WHO classification of these tumors (5th edition expected to be published within a year from now!). When shaping a WHO classification, one may indeed follow such a more progressive approach (i.e. introduce the latest tools and findings and include e.g. the most refined subclassification of non-WNT/non-SHH medulloblastomas), or be somewhat more conservative in an attempt to better preserve long-term correlations, to avoid major disruption of patient management, and/or because of limited availability of (often expensive) diagnostic tools. Obviously, finding the right balance is key, as there are many ‘strings attached’ (Fig. 3).
Fig. 3. ‘Strings attached’ to designing a next WHO classification of (CNS) tumors. Next to diagnostic and prognostic markers, clinical neuropathologists are increasingly asked to perform testing for predictive biomarkers, e.g. for assessment of the likelihood of response of gliomas to particular immunotherapeutic approaches. Only a limited number of newly diagnosed gliomas is reported to have an (occasionally inherited) mismatch repair (MMR) defect and/or a ‘hypermutator’ phenotype. Recurrent gliomas more often show such a phenotype, especially so after alkylating chemotherapy [8]. Assuming that the hypermutator status leads to an increase in expression of neoantigens, these gliomas/glioblastomas with a hypermutator phenotype could be good candidates for immune checkpoint blockade. Unequivocal scoring of immunohistochemical staining for PD1/PD-L1 (surface proteins on tumor-infiltrating lymphocytes and tumor cells, respectively, and involved in suppression of the immune system) can be challenging. Alternatively, MMR deficiency, microsatellite instability (MSI), DNA polymerase epsilon (POLE) mutations, and high tumor mutational load (TML) are being used in this context, but assessment of these biomarkers can be challenging as well. For example, MMR gene (MLH1, MSH2, MSH6, and/or PMS2) mutations often, but not always lead to lack of immunostaining of tumor cell nuclei for the corresponding protein(s). Also, the techniques and thresholds to be used for optimal assessment of high TML are not yet settled. While for non-small cell lung carcinoma ≥10 mutations per Mb is considered as high TML, it is unclear if this threshold should be used for gliomas as well [56, 57]. Of course, identification and optimization of biomarkers for response to immunotherapy only makes sense if at least some patients can be expected to significantly benefit from that particular therapy. Furthermore, the studies reported under Topic 9 (‘Personalized (neo)antigen vaccination therapy for glioblastoma’) indicate that biomarker discovery in this context is a moving target. In conclusion, ‘the times they are a-changin’ and one can expect that there is much more change to come. Liquid biopsy-based diagnoses may emerge soon as a clinically helpful source of information. Furthermore, a very recently published study showed that the use of artificial intelligence outperformed radiologists in the mammogram-based prediction of breast cancer [58]. Similarly, deep learning/machine learning approaches may increasingly be used for automated, histology-based brain tumor classification and/or for predicting molecular markers based on MRI or other neuro-imaging modalities [59, 60]. Such developments could have significant impact on the ‘core business’ of the (neuro)pathologist. However, to quote the Editor-in-Chief of Free Neuropathology: ‘As long as neuropathology continues to be creative and innovative at the forefront of classifying and diagnosing neurological disorders and revealing their molecular pathogenesis …, its future will be bright’ [52]. References 1. Filipski, K., et al., Lack of H3K27 trimethylation is associated with 1p/19q codeletion in diffuse gliomas. Acta Neuropathol, 2019. 138(2): p. 331-334. 2. Sievers, P., et al., Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol, 2019. 138(3): p. 497-504. 3. Hou, Y., et al., Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol, 2019. 137(5): p. 837-846. 4. Sharma, T., et al., Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol, 2019. 138(2): p. 309-326. 5. Vladoiu, M.C., et al., Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature, 2019. 572(7767): p. 67-73. 6. Hovestadt, V., et al., Resolving medulloblastoma cellular architecture by single-cell genomics. Nature, 2019. 572(7767): p. 74-79. 7. Jessa, S., et al., Stalled developmental programs at the root of pediatric brain tumors. Nat Genet, 2019. 51(12): p. 1702-1713. 8. Barthel, F.P., et al., Longitudinal molecular trajectories of diffuse glioma in adults. Nature, 2019. 576(7785): p. 112-120. 9. Wang, J., et al., Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci, 2019. 22(1): p. 91-105. 10. Venkatesh, H.S., et al., Electrical and synaptic integration of glioma into neural circuits. Nature, 2019. 573(7775): p. 539-545. 11. Venkataramani, V., et al., Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature, 2019. 573(7775): p. 532-538. 12. Zeng, Q., et al., Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature, 2019. 573(7775): p. 526-531. 13. Dirkse, A., et al., Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun, 2019. 10(1): p. 1787. 14. Keskin, D.B., et al., Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature, 2019. 565(7738): p. 234-239. 15. Hilf, N., et al., Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature, 2019. 565(7738): p. 240-245. 16. Miller, A.M., et al., Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature, 2019. 565(7741): p. 654-658. 17. Pan, C., et al., Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol, 2019. 137(2): p. 297-306. 18. Paramasivam, N., et al., Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol, 2019. 138(2): p. 295-308. 19. Wesseling, P., M. van den Bent, and A. Perry, Oligodendroglioma: pathology, molecular mechanisms and markers. Acta Neuropathol, 2015. 129(6): p. 809-27. 20. Schaefer, I.M., A. Minkovsky, and J.L. Hornick, H3K27me3 immunohistochemistry highlights the inactivated X chromosome (Xi) and predicts sex in non-neoplastic tissues. Histopathology, 2016. 69(4): p. 702-4. 21. Panwalkar, P., et al., Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol, 2017. 134(5): p. 705-714. 22. Cleven, A.H., et al., Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol, 2016. 29(6): p. 582-90. 23. Katz, L.M., et al., Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol, 2018. 135(6): p. 955-963. 24. Capper, D., et al., DNA methylation-based classification of central nervous system tumours. Nature, 2018. 555(7697): p. 469-474. 25. Capper, D., et al., Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol, 2018. 136(2): p. 181-210. 26. Jaunmuktane, Z., et al., Methylation array profiling of adult brain tumours: diagnostic outcomes in a large, single centre. Acta Neuropathol Commun, 2019. 7(1): p. 24. 27. Pickles, J.C., et al., DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc Health, 2020. 4(2): p. 121-130. 28. Perez, E. and D. Capper, DNA-Methylation-based Classification of Paediatric Brain Tumours. Neuropathol Appl Neurobiol, 2020. 29. Priesterbach-Ackley, L.P., et al., Brain tumour diagnostics using a DNA methylation-based classifier as a diagnostic support tool. Neuropathol Appl Neurobiol, 2020. 30. Schwalbe, E.C., et al., Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol, 2017. 18(7): p. 958-971. 31. Cavalli, F.M.G., et al., Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell, 2017. 31(6): p. 737-754 e6. 32. Northcott, P.A., et al., The whole-genome landscape of medulloblastoma subtypes. Nature, 2017. 547(7663): p. 311-317. 33. Gibson, P., et al., Subtypes of medulloblastoma have distinct developmental origins. Nature, 2010. 468(7327): p. 1095-9. 34. Cancer Genome Atlas Research, N., Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 2008. 455(7216): p. 1061-8. 35. Brennan, C.W., et al., The somatic genomic landscape of glioblastoma. Cell, 2013. 155(2): p. 462-77. 36. Cancer Genome Atlas Research, N., et al., Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med, 2015. 372(26): p. 2481-98. 37. Ceccarelli, M., et al., Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell, 2016. 164(3): p. 550-63. 38. Johnson, B.E., et al., Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science, 2014. 343(6167): p. 189-193. 39. van Thuijl, H.F., et al., Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol, 2015. 129(4): p. 597-607. 40. Shirahata, M., et al., Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol, 2018. 136(1): p. 153-166. 41. Claes, A., A.J. Idema, and P. Wesseling, Diffuse glioma growth: a guerilla war. Acta Neuropathol, 2007. 114(5): p. 443-58. 42. Venkatesh, H.S., et al., Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell, 2015. 161(4): p. 803-16. 43. Venkatesh, H.S., et al., Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature, 2017. 549(7673): p. 533-537. 44. Osswald, M., et al., Brain tumour cells interconnect to a functional and resistant network. Nature, 2015. 528(7580): p. 93-8. 45. Gimple, R.C., et al., Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev, 2019. 33(11-12): p. 591-609. 46. Zhao, J., et al., Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med, 2019. 25(3): p. 462-469. 47. Cloughesy, T.F., et al., Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med, 2019. 25(3): p. 477-486. 48. Best, M.G., et al., Liquid biopsies in patients with diffuse glioma. Acta Neuropathol, 2015. 129(6): p. 849-65. 49. van der Pol, Y. and F. Mouliere, Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell, 2019. 36(4): p. 350-368. 50. Kristensen, B.W., et al., Molecular pathology of tumors of the central nervous system. Ann Oncol, 2019. 30(8): p. 1265-1278. 51. Priesterbach-Ackley, L.P., et al., Molecular tools for the pathologic diagnosis of central nervous system tumors. Neurooncol Pract, 2019. 6(1): p. 4-16. 52. Paulus, W., HEIRECA! The HEIdelberg REvolution of CAncer classification and what it means for neurooncology and neuropathology. Acta Neuropathol, 2018. 136(2): p. 177-179. 53. Wesseling, P., The ABCs of molecular diagnostic testing of CNS tumors: acceptance, benefits, costs. Neuro Oncol, 2019. 21(5): p. 559-561. 54. Karimi, S., et al., The central nervous system tumor methylation classifier changes neuro-oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin Epigenetics, 2019. 11(1): p. 185. 55. Louis, D.N., et al., cIMPACT-NOW: a practical summary of diagnostic points from Round 1 updates. Brain Pathol, 2019. 29(4): p. 469-472. 56. Hodges, T.R., et al., Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol, 2017. 19(8): p. 1047-1057. 57. Tan, A.C., A.B. Heimberger, and M. Khasraw, Immune Checkpoint Inhibitors in Gliomas. Curr Oncol Rep, 2017. 19(4): p. 23. 58. McKinney, S.M., et al., International evaluation of an AI system for breast cancer screening. Nature, 2020. 577(7788): p. 89-94. 59. Ker, J., et al., Automated brain histology classification using machine learning. J Clin Neurosci, 2019. 66: p. 239-245. 60. Korfiatis, P. and B. Erickson, Deep learning can see the unseeable: predicting molecular markers from MRI of brain gliomas. Clin Radiol, 2019. 74(5): p. 367-373. 61. Gritsenko, P.G., et al., p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat Cell Biol, 2020. 22(1): p. 97-107.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |