|
|
|
>Free Neuropathology 1:4 (2020) |
|
Review |
|
Top ten discoveries of the year: Neuromuscular disease |
|
Marta Margeta |
|
Department of Pathology, University of California, San Francisco, CA, USA |
|
Corresponding author: |
|
Submitted: 15 January 2020 Accepted: 21 January 2020 Published: 23 January 2020 |
|
Keywords: Anterior horn disease, Congenital myopathy, Inflammatory myopathy, Muscular dystrophy, Neuropathy, Radiculopathy, RNA-seq, Gene therapy |
|
Abstract This review highlights ten important advances in the neuromuscular disease field that either were first reported in 2019, or have reached a broad consensus during that year. The overarching topics include (i) new / emerging diseases; (ii) advances in understanding of disease etiology and pathogenesis; (iii) diagnostic advances; and (iv) therapeutic advances. Within this broad framework, the individual disease entities that are discussed in more detail include myoglobinopathy, POPDC3-mutated limb-girdle muscular dystrophy, neuromuscular adverse events associated with the immune checkpoint inhibition therapy, neuroglial stem cell-derived inflammatory pseudotumor of the spinal cord and spinal cord roots, acute flaccid myelitis, congenital myopathies, idiopathic inflammatory myopathies (with particular emphasis on immune-mediated necrotizing myopathies and sporadic inclusion body myositis), spinal muscular atrophy, and Duchenne muscular dystrophy. In addition, the review highlights several diagnostic advances (such as diagnostic RNA sequencing and development of digital diagnostic tools) that will likely have a significant impact on the overall neuromuscular disease field going forward. Abbreviations AAV, adeno-associated virus; AFM, acute flaccid myelitis; AI, artificial intelligence; ASM, antisynthetase syndrome-associated myositis; ASO, antisense oligonucleotide; CASA, chaperone-assisted selective autophagy; DM, dermatomyositis; DMD, Duchenne muscular dystrophy; EMA, European Medicines Agency; EV, enterovirus; FDA, Federal Drug Administration; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; IFN, interferon; IIM, idiopathic inflammatory myopathy; IMNM, immune-mediated necrotizing myopathy; irAE, immune-related adverse event secondary to immune checkpoint inhibition; irMG, myasthenia gravis secondary to immune checkpoint inhibition; irMyositis, inflammatory myopathy secondary to immune checkpoint inhibition; irNeuropathy, immune-mediated neuropathy secondary to immune checkpoint inhibition; MHC, major histocompatibility complex; mNGS, metagenomic next generation sequencing; MSA, myositis-specific antibodies; MxA, myxovirus resistance protein A; NM, nemaline myopathy; NMD, neuromuscular disease; n-SCIPT, neuroglial stem cell-derived inflammatory pseudotumor; PM, polymyositis; RNA-seq, transcriptome analysis (RNA sequencing); sIBM, sporadic inclusion body myositis; SMA, spinal muscular atrophy; SRP, signal recognition particle; TEMRA cells, terminally differentiated effector memory T cells; WES, whole exome sequencing Introduction 2019 was an exciting year for the neuromuscular disease (NMD) field, with important developments on multiple fronts and in many different disease categories. In this review paper, I will summarize ten 2019 NMD discoveries that I consider to be most important and/or interesting; they are grouped in four different “discovery clusters” and listed in no particular order. (Like most classifications, this one is not perfect – a few discoveries included in the “etiology/pathogenesis cluster” have direct implications for diagnostics and/or treatment of NMDs – but I hope it will improve the overall readability of the review.) Newly defined / emerging neuromuscular diseases 1. Newly defined genetic diseases Even as our understanding of the NMD genetics inches closer to completion, new genetic NMDs continue to be defined; in this review, I will summarize key features of two particularly interesting entities that were first characterized in 2019. Myoglobinopathy is caused by the c.292C>T, p.His98Tyr mutation in myoglobin, a small cytoplasmic hemoprotein that buffers intracellular O2 concentration and regulates the intracellular redox potential in cardiomyocytes and type 1 muscle fibers (Olive et al., 2019). Through an effort that involved two research teams on two different continents, this mutation was detected in 14 individuals from six different families, all of whom were initially identified based on the unique clinicopathologic features of their disease. Clinically, myoglobinopathy presents with a weakness of the proximal lower limb and axial muscles; the symptoms start in the fourth to fifth decade and are slowly progressive, ultimately involving distal lower limb, upper limb, and respiratory muscles. Creatine kinase levels are usually normal, while cardiac involvement is clinically apparent in approximately half of the affected individuals. Pathologically, all muscle biopsies (as well as the hearts from two individuals who underwent autopsy) showed characteristic sarcoplasmic bodies that are brown on H&E stain, red on modified Gomori trichrome stain, and highly electron dense on electron microscopy (Fig. 1); interestingly, these sarcoplasmic inclusions rarely contain myoglobin and seem to be a product of increased lipid oxidation. (Late stage biopsies also show chronic myopathic changes and autophagic impairment including rimmed vacuoles.) Although additional work will be required to fully elucidate the molecular mechanisms underlying this newly defined disease, the initial biochemical experiments suggest that mutant myoglobin is more prone to the loss of the heme moiety and is associated with elevated intracellular superoxide levels, suggesting that impaired redox balance plays a role in the pathogenesis of this disease.
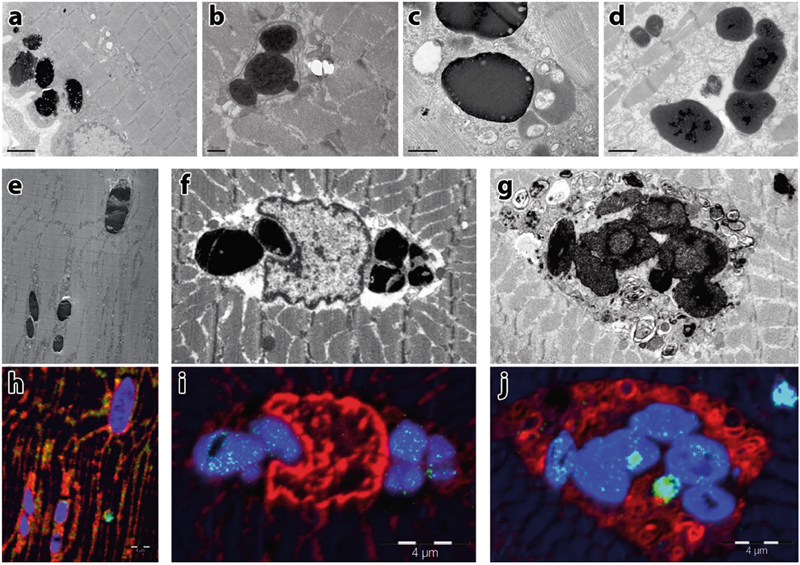
Figure 1. Ultrastructural characterization of sarcoplasmic bodies, the morphological hallmark of myoglobinopathy: Electron micrographs (a–d) show collections of highly electron-dense bodies with some less dense material at their periphery. The sarcoplasmic bodies are seen under the sarcolemma (a), often next to the nuclei. Some sarcoplasmic bodies are surrounded by a membrane (b). Sarcoplasmic bodies of different electron densities are seen near several vesicular structures (c) and were observed in the cardiac muscle obtained post mortem from one of the affected individuals (d). Electron micrographs (e–g) and the corresponding Nanoscale Secondary Ion Mass Spectrometry images (h–j). Blue indicates sulfur (32S), red phosphorus (31P), and green iron (56Fe), respectively. Sarcoplasmic bodies are seen interspersed between the myofibrils (e), next to nuclei (f), or inside an autophagic vacuole (g). Note the high-sulfur signal in the sarcoplasmic bodies (h–j), and the iron signal (green dots within the sarcoplasmic bodies in i, j). Scale bar in a = 2 µm, b = 5 µm, c = 0.5 µm, d = 1 µm, h–j = 4 µm. [This figure and its legend were adopted from Figure 5 in (Olive et al., 2019); this use is permitted under the Creative Commons Attribution 4.0 International License.] A different, purely genetic approach was used to define a new autosomal recessive form of limb-girdle muscular dystrophy that is caused by biallelic mutations in POPDC3 gene: 5 affected individuals from 3 different families were identified via exome sequencing of a cohort of 1500 patients with presumed genetic limb-girdle weakness and elevated creatine kinase levels; their muscle biopsies showed dystrophic features of variable severity (Vissing et al., 2019). POPDC3 (Popeye domain containing 3) gene encodes a membrane protein that regulates cAMP signaling, is highly expressed in skeletal and cardiac muscles, and has not yet been associated with any human disease. Interestingly, patients with POPDC3 mutations do not show a cardiac phenotype, while mutations in two other members of the same gene family (POPDC1 and POPDC2) cause a cardiac phenotype (generally a conduction disorder) accompanied by a variable skeletal muscle involvement (De Ridder et al., 2019; Vissing et al., 2019). The pathogenesis of this new disorder remains to be elucidated; however, initial studies suggest a loss-of-function mechanism that leads to altered cAMP modulation. 2. Emerging therapy-related diseases Introduction of new therapies has led to the emergence of unusual or completely novel NMD phenotypes; in 2019, two new NMD categories, previously described only in individual case reports or small case series, have been more thoroughly defined. Immune-checkpoint inhibitor therapy has revolutionized treatment of patients with advanced cancers, but is often associated with immune-related adverse events (irAEs) that most commonly affect the skin, GI tract, and endocrine glands, as well as the PNS. Neuromuscular irAEs can be severe and include myasthenia gravis (irMG), myopathy (irMyositis), and peripheral neuropathy (irNeuropathy; most commonly acute demyelinating polyradiculoneuropathy) (Johansen et al., 2019; Mohn et al., 2019; Psimaras et al., 2019). While these neuromuscular irAEs share some commonalties with their sporadic counterparts, several unique features have emerged. For example, up to 25% of the patients show more than one neuromuscular irAE, with the combination of irMG and irMyositis encountered most often (Johansen et al., 2019). Furthermore, irMG is more likely to be negative for AChR antibodies than idiopathic MG, while irMyositis is more likely to involve oculomotor and bulbar muscles than any of idiopathic inflammatory myopathies (IIMs); these features make clinical distinction between irMG and irMyositis difficult in many cases (Johansen et al., 2019; Mohn et al., 2019). In addition, irMyositis often involves axial muscles, while IIMs do not (Touat et al., 2018). Finally, a significant fraction of patients with irMyositis shows cardiac involvement, which is not typically seen in IIM patients and has a poor prognosis (Anquetil et al., 2018; Johansen et al., 2019; Mohn et al., 2019). Interestingly, while the rates of irNeuropathies (as well as the overall irAE rates) are higher in patients treated with ipilimubab (the anti-CTLA-4 antibody), irMG and irMyositis are particularly common in patients treated with nivolumab or pembrolizumab (the PD-1 blocking antibodies) (Mohn et al., 2019). The pathogenesis of neuromuscular irAEs is incompletely understood, but is thought to involve both type IV (T cell-mediated) and type II (antibody-mediated) immune mechanisms (Psimaras et al., 2019). On a tissue level, irMyositis can take multiple forms but the most commonly observed pattern [perimysial foci of CD8+ T cells and CD68+ macrophages that are associated with necrosis of adjacent muscle fibers; Touat et al. (2018) and Fig. 2] is distinct from the patterns seen in IIMs. Interestingly, in some cases identical clonal CD8+ cells have been detected within the cancer tissue, skeletal muscle, and myocardium, suggesting that cross-reactivity between cancer cells and myocytes plays at least some role in the pathogenesis of irMyositis (Psimaras et al., 2019).
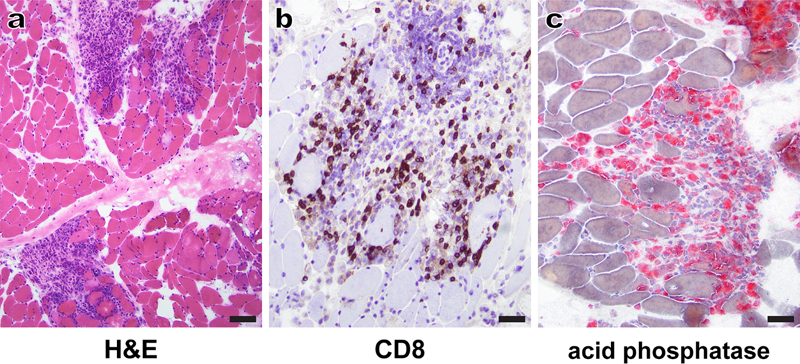
Figure 2. Myositis secondary to therapy with immune checkpoint inhibitors (irMyositis): A representative H&E-stained cryosection (a) shows patches of dense perimysial inflammation that focally extend into the endomysium and abut clusters of degenerating/regenerating muscle fibers; away from the inflammatory foci, muscle fibers have a relatively normal appearance. Immunohistochemistry for CD8 (b) and histochemistry for acid phosphatase (c) show that the inflammatory infiltrate largely consists of cytotoxic T cells and macrophages, respectively. The patient was treated with pembrolizumab and presented with evidence of myocarditis as well as limb and bulbar weakness approximately 6 weeks following the last antibody dose. Scale bars: a, 100 µm; b-c, 50 µm. In recent years, stem cell treatments have also risen in popularity, although they have no documented health benefits and are – at least for now – completely unregulated. In 2019, it was recognized that one relatively common form of stem cell treatment, intrathecal injection of allogeneic stem cells, can lead to development of distinctive lesions of the lumbosacral spinal cord and spinal cord nerve roots. These lesions, named neuroglial stem cell-derived inflammatory pseudotumors [n-SCIPTs; (Sloan et al., 2019)], can clinically mimic inflammatory and neoplastic disease processes and usually consist of a haphazardly organized neuroglial tissue that resembles a low-grade neuroglial tumor (Fig. 3); typically, there is at least focal inflammatory infiltrate, which likely represents a host immune response to foreign antigens. While it seems likely that direct injection of allogeneic stem cells into other parts of the body could lead to development of stem-cell derived pseudotumors with non-neuroglial differentiation, only neuroglial SCIPTs have been reported to date (Sloan et al., 2019). From a diagnostic perspective, it is critical to be aware of the n-SCIPT existence, so that these lesions (which at the moment seem to be best handled through watchful waiting) are distinguished from the spinal cord tumors that would generally warrant a more aggressive treatment. Advances in understanding of etiology and pathogenesis of neuromuscular diseases 3. Acute flaccid myelitis: Mounting evidence for causation by non-polio enteroviruses Acute flaccid myelitis (AFM) is a clinical syndrome characterized by flaccid limb weakness that develops acutely and has a lower motor neuron pattern; the current diagnostic criteria also include spinal cord imaging findings suggestive of anterior myelitis (Messacar et al., 2018). The most common form of AFM used to be poliomyelitis, which is caused by poliovirus [a member of the En terovirus (EV) genus]; as a result, AFM became rare worldwide after introduction of the anti-polio vaccine in the 1950s. A small number of AFM cases in the period from 1970-90 was attributed to infection with EV-A71 and a few other viruses (Dyda et al., 2018; Messacar et al., 2018). Since 2012, however, AFM has started to increase in incidence. The current form of this disease, first documented in California, is characterized by limb paralysis that occurs 5-7 days after a flu-like respiratory illness; it primarily affects young children (3-9 years of age) and peaks in incidence every other year during the summer/fall season. The largest number of confirmed cases (>500 to date) has been reported in the US, with sporadic cases confirmed throughout the world (Schubert et al., 2019). In 2014, the peak of AFM incidence has shown a temporal and geographic overlap with the outbreak of EV-D68 infection, raising the possibility that EV-D68 (which rarely caused human disease prior to 2008) might be the cause of recent AFM outbreaks (Dyda et al., 2018). Indeed, EV-D68 infection meets many of the Bradford-Hill criteria (nine principles applied to examine causality) that are required to establish it as a cause of AFM (Dyda et al., 2018; Messacar et al., 2018). In particular, AFM-like disease can be recapitulated in a mouse model through infection with a 2014 strain (but not the originally isolated 1962 strain) of EV-D68, suggesting that neurotropism might have emerged only recently in EV-D68 evolution (Hixon et al., 2017). However, 2016 AFM cases have not shown a clear association with an EV-D68 outbreak, raising doubts about the causal association between EV-D68 and AFM (Dyda et al., 2018). Moreover, EV has not been detected in the respiratory or GI specimens from more than half of AFM children, and EV RNA is very rarely detected in the CSF of AFM patients (Schubert et al., 2019); thus, concerns have persisted that recent AFM outbreaks might be caused by a still unidentified pathogen or an autoimmune mechanism. The 2019 study by Schubert et al. has used a multi-pronged approach [metagenomic ultra-deep next generation sequencing (mNGS) of the CSF to detect the pathogen-derived nucleic acids, and antiviral antibody detection in the CSF using VirScan phage display library and EV ELISA) to analyze 42 AFM cases (covering all outbreaks from 2012-2018) and 58 neurologic disease controls. Except for one already known EV-D68-positive case, mNGS failed to discover any CSF pathogens in either group. However, VirScan and EV ELISA revealed significantly higher levels of anti-EV antibodies in cases compared to controls, providing a strong support for EVs as causative agents of all recent AFM outbreaks. Interestingly, CSF from AFM cases commonly contained antibodies targeting more than one EV; thus, viral detection in the spinal cord tissue will likely be required to definitively establish which EV species can cause AFM. In addition, further work will be required to determine the sensitivity and specificity of CSF EV serology in AFM diagnosis (Schubert et al., 2019).
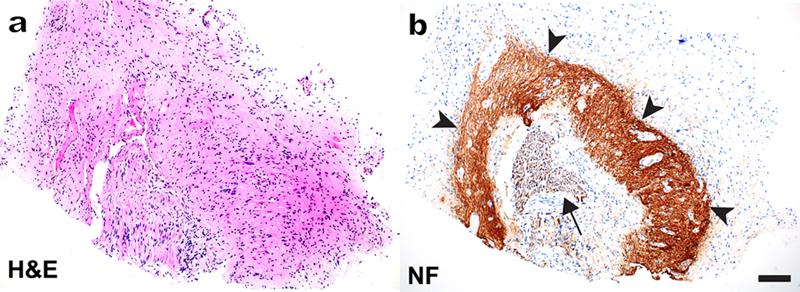
Figure 3. Neuroglial stem cell-derived inflammatory pseudotumor (n-SCIPT): A representative H&E-stained paraffin section (a) shows a spinal cord nerve root surrounded by haphazardly organized but otherwise mature-appearing neuroglial tissue. Neurofilament immunohistochemistry (b) highlights intensely stained but disorganized nerve cell processes (arrowheads) that surround the centrally located nerve root (arrow); the tissue at the periphery of the specimen shows glial differentiation and is neurofilament-negative. Chronic inflammatory infiltrate was apparent on other sections; for additional images and clinical history details, see the original publication (Sloan et al., 2019). Scale bar, 100 µm. 4. Congenital myopathies: New pathogenetic mechanisms Congenital myopathies are a group of genetically and clinically heterogeneous disorders that typically present early in life with weakness / hypotonia and delayed motor development, and are subsequently characterized by a stable or slowly progressive disease course. Histopathologic findings are variable and include type 1 fiber predominance and atrophy, cores and/or minicores, mislocalization of intracellular organelles (including nuclei), and various sarcoplasmic inclusions (such as nemaline rods, caps, and cytoplasmic bodies). The well-established pathogenetic mechanisms include impairments of calcium homeostasis, excitation-contraction coupling, sarcomeric filament assembly, force generation, and membrane remodeling. Excitingly, this mechanistic spectrum has been significantly widened by the work published in 2019. Some of the newly discovered mechanisms include (i) cell cycle acceleration accompanied by reduced cell growth (Villar-Quiles et al., 2019), (ii) impaired redox balance (Lornage et al., 2019), (iii) altered muscle kinetics due to impaired sarcomere relaxation (de Winter et al., 2020), and (iv) nuclear dysfunction secondary to impaired contractility (Ross et al., 2019); these last two studies are particularly groundbreaking and will therefore be described in a greater detail. Nemaline myopathy (NM) can be caused by mutations in at least 13 genes, the great majority of which encode proteins that either form thin filaments or are associated with these filaments. A recent tour-de-force study by de Winter et al. (2020; advanced electronic publication in 2019) has established that KBTBD13 (kelch repeat and BTB domain containing 13), the mutations in which cause NEM6 subtype of NM, is also an actin-binding protein; when mutated, KBTBD13 impairs sarcomere relaxation by altering the structure of thin filaments. Specifically, thin filaments are more tightly wound and stiffer than normal when they are bound to mutant KBTBD13R408C; this change in the thin filament compliance disrupts the positive feedback loop that is required for thin-thick filament detachment, resulting in slowing of muscle relaxation that clinically characterizes NEM6. Given the 8:1 stoichiometry of KBTBD13-actin binding, which is close to the 7:1 stoichiometry between troponin / tropomyosin and actin, it is possible that KBTBD13 acts in concert with those well-known sarcomeric regulatory proteins to fine-tune muscle contraction kinetics. Interestingly, deletion of KBTBD13 in mice does not lead to slower muscle relaxation, suggesting that KBTBD13R408C is a gain-of-function mutation. It remains to be shown whether other mutations in this gene lead to similar functional deficits and to elucidate how defects in muscle relaxation ultimately lead to production of nemaline rods (which are recapitulated in the mouse KBTBD13R408C knock-in model). In 2019, we have also learned that impaired contractility and abnormal cytoskeletal organization produce secondary nuclear deficits, which structurally – and possibly functionally – mimic nuclear abnormalities seen in patients with mutations in the nuclear envelope proteins, such as lamins, nesprins, and emerin (Ross et al., 2019). Abnormal spacing of myonuclei, nuclear envelope defects, and chromatin alterations were present not only in the skeletal muscle from patients with two different genetic forms of NM and NM model mice, but also in skeletal muscle from patients with acquired (immune-mediated) forms of NM, underscoring that these nuclear abnormalities are secondary to impaired contractility rather than a direct consequence of NM mutations. In addition, muscle fibers from both NM patients and NM model mice showed extensive cytoskeletal disruption (disorganized microtubules, mislocalized intermediate filaments composed of desmin, and altered cortical microfilaments composed of actin); given that treatment with microtubule-disrupting agents was shown to alter nuclear shape in vitro, these cytoskeletal alterations likely also contribute to nuclear deficits seen in the NM muscle. Indeed, we have recently observed very similar nuclear defects in a patient with a desmin mutation (Fig. 4); thus, secondary nuclear defects (and consequent gene expression changes) may be an important but overlooked cause of muscle dysfunction that is more widespread than currently recognized.
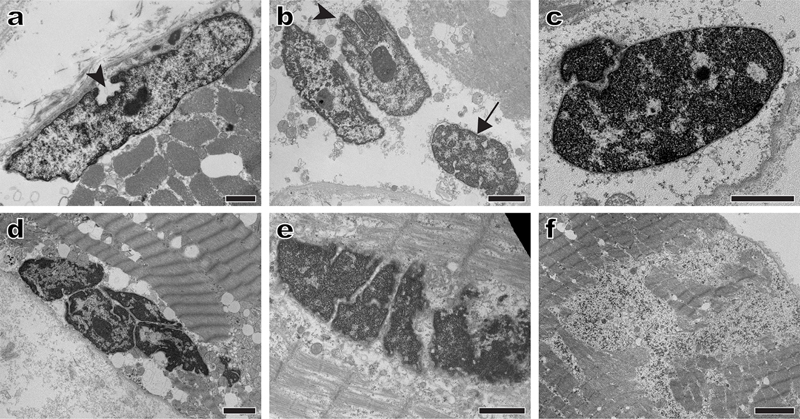
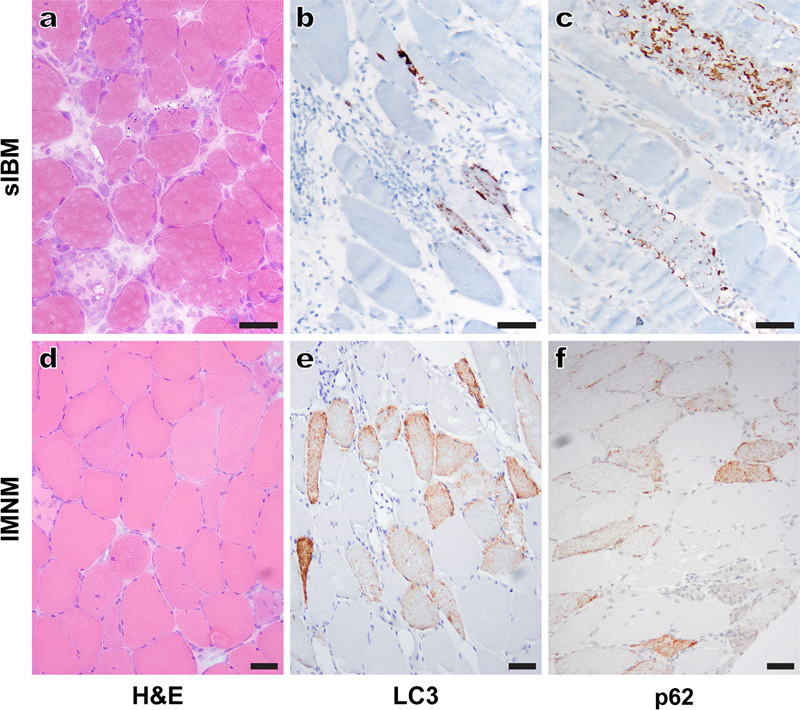
Figure 4. Nuclear abnormalities in a case of myofibrillar myopathy caused by a desmin mutation: Representative electron micrographs show marked nuclear abnormalities (a-e) including invaginations of the nuclear membrane (a and b, arrowheads), abnormally dense chromatin (the nucleus highlighted by the arrow in b, as well as nuclei in c and d), and nuclear fragmentation (c-e). Granulofilamentous material (f), a clue to the correct diagnosis, was seen only focally. Subsequent genetic testing revealed a heterozygous DES gene mutation [c.1237_1239delGAG (p.E413del)]. Scale bars: a and e, 1 µm; b-d, 2 µm; f, 4 µm. 5. Sporadic inclusion body myositis: The role of terminally differentiated cytotoxic T cells Sporadic inclusion body myositis (sIBM) is the most common skeletal myopathy affecting people over the age of 50; it typically presents with a weakness of the quadriceps and deep finger flexor muscles, has a slowly progressive course resulting in eventual loss of mobility, and is currently without effective therapy. Pathologically, sIBM shows both inflammatory and myodegenerative features (Fig. 5a); together with a lack of response to immunosuppressive treatment, this “dual” nature of sIBM pathology has led to a long-standing disagreement about the role of inflammation in sIBM pathogenesis.Three studies published in 2019 (Dzangue-Tchoupou et al., 2019; Greenberg et al., 2019; Knauss et al., 2019) will likely break this impasse by providing a strong support for the autoimmune nature of sIBM: starting with different questions and using different experimental approaches, these studies have demonstrated that terminally differentiated effector memory T cells (so-called TEMRA cells) are enriched in sIBM muscle biopsies (Greenberg et al., 2019; Knauss et al., 2019) as well as peripheral blood of sIBM patients (Dzangue-Tchoupou et al., 2019), and that the presence of these cells is fairly unique for sIBM when compared to other IIMs [although similar cells were also seen in biopsies from two patients with severe, treatment-resistant irMyositis (Knauss et al., 2019)]. TEMRA cells are positive for CD8, T-bet (a transcription factor that drives differentiation of naïve T cells into cytotoxic T lymphocytes), CD57 (a marker of replicative senescence), and KLRG1 (killer cell lectin-like receptor G1; a marker of T cell exhaustion). They contain high concentration of cytotoxic enzymes (granzymes and perforin) that likely directly cause muscle fiber damage, are known to increase in peripheral blood with age, and are resistant to corticosteroids and drugs that target actively dividing cells (such as methotrexate); the latter two characteristics likely provide at least partial explanation for the age dependence and resistance to immunosuppression that are characteristic of sIBM.
Figure 5. Distinct patterns of LC3 and p62 immunostaining in sporadic inclusion body myositis (sIBM) and immune-mediated necrotizing myopathy (IMNM): A typical sIBM case (a-c) shows inflammatory and myodegenerative changes on H&E staining (a) and dense, largely subsarcolemmal protein aggregates and vacuoles on LC3 (b) and p62 (c) immunostaining. In contrast, an IMNM case (d-f) shows randomly distributed degenerating/regenerating fibers and the absence of significant inflammation on H&E (d) and multiple fibers with diffuse, finely punctate staining on LC3 (e) and p62 (f) immunostaining. For both cases, H&E stain was performed on cryosections, while LC3 and p62 immunohistochemistries were performed on formalin-fixed, paraffin-embedded tissue sections. Scale bars, 50 µm. In addition to providing insight into sIBM pathogenesis, these studies have important implications for the sIBM diagnosis and treatment. Of particular interest, the study by Greenberg at al. (2019) included 10 samples from patients diagnosed with polymyositis (PM), an IIM that was historically thought to share the sIBM inflammatory profile but lack its myodegenerative phenotype and its refractoriness to therapy. The existence of PM as a distinct IIM subtype has recently been called into question, given that most patients diagnosed with PM based on the 1975 Bohan & Peter criteria can be reclassified as one of the other IIMs [early stage sIBM, immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome-associated myositis (ASM), or dermatomyositis (DM)] using the current clinicoseropathologic criteria (Allenbach et al., 2017; Tanboon and Nishino, 2019). However, Greenberg and co-authors have demonstrated an important difference between their “PM” and sIBM samples: while CD8+ lymphocytes invading muscle fibers in sIBM biopsies were abundant and CD57-positive, the CD8+ T lymphocytes invading muscle fibers in “PM” biopsies were sparse and CD57-negative. Additional work will be required to establish whether pathologically diagnosed PM is a distinct (albeit rare) disease entity or whether it corresponds to an early stage of sIBM that lacks senescent, CD57+KLRG1+ TEMRA cells and thus remains responsive to immunosuppressive therapy. Either way, these findings raise a possibility that the presence of CD8+CD57+KLRG1+ T cells in an IIM biopsy could serve as a prognostic marker of resistance to the currently available immunosuppressive therapies, a clinically important application that warrants future study. In addition, expression of KLRG1 by sIBM TEMRA cells may have therapeutic implications: given that this protein is not expressed by central memory T cells and regulatory T cells, it could be used as a basis for development of a future sIBM therapeutic (Greenberg et al., 2019). 6. Immune-mediated necrotizing myopathies: Mechanisms of cell injury mmune-mediated necrotizing myopathies (IMNMs) are a unique class of IIMs for which the consensus clinicoseropathologic criteria were defined at a workshop held in 2016 (Allenbach et al., 2018). IMNMs have been linked to autoantibodies against signal recognition particle (SRP) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), but approximately one third of cases is seronegative and requires histopathologic diagnosis (Bergua et al., 2019). Regardless of the serologic subtype, pathologic features of IMNM include randomly distributed necrotic and regenerating muscle fibers (Fig. 5d), variable MHC-1 upregulation, sarcolemmal complement deposition, and a scant to absent lymphocytic infiltrate. Previous work has shown that in vitro treatment with either anti-SRP or anti-HMGCR antibodies leads to impaired myoblast fusion and muscle fiber atrophy (Arouche-Delaperche et al., 2017), suggesting that these autoantibodies play a causative role in IMNM pathogenesis. A study from the same group published in 2019 has taken the field a step further by demonstrating the causative effect of these two antibodies in vivo: mice treated with either anti-SRP+ or anti-HMGCR+ patient-derived plasma develop weakness and necrotizing myopathy that closely resembles human IMNM (Bergua et al., 2019). In a further agreement with human disease, muscle deficiency is worse following treatment with anti-SRP than with anti-HMGCR antibodies, but both antisera require functional complement pathway to produce the disease phenotype. Taken together, these studies provide a conclusive evidence for the key role of type II (antibody- and complement-mediated) immune mechanisms in IMNM pathogenesis. Interestingly, target antigens of the two known IMNM autoantibodies (the signal peptide-binding 54 kDa subunit of the SRP ribonucleoprotein complex and HGMCR, the rate-limiting enzyme in the cholesterol biosynthetic pathway) are both expressed in the ER of all cell types (Bergua et al., 2019). Given the ubiquitous expression of these antigens, it remains unclear why anti-SRP and anti-HMGCR antibodies selectively target skeletal muscle in both humans and mice. However, the ER localization of both autoantibody targets may be significant for IMNM pathogenesis: a 2019 study has shown that human IMNM muscle biopsies of any serotype show increased ER stress, activation of the unfolded protein response, and upregulation of the chaperone-assisted selective autophagy (CASA) pathway (Fischer et al., 2019). Notably, activation of the CASA pathway in IMNMs is associated with a unique pattern of immunostaining for autophagy-associated proteins LC3 and p62/SQSTM1: instead of the coarse LC3- and p62-positive vacuoles and protein aggregates seen in autophagic vacuolar myopathies and sIBM [Figs. 5b and 5c; also reviewed in (Margeta, 2019)], IMNM biopsies show diffusely punctate and often quite strong staining of a variable subset of non-necrotic muscle fibers (Figs. 5e and 5f). While it remains to be shown how antibody-mediated cell injury leads to activation of the CASA pathway [the impairment of which plays a role in the pathogenesis of myofibrillar myopathies; reviewed in (Margeta, 2019)], this unique LC3- and p62-staining pattern is likely to prove useful in diagnostically challenging IMNM cases; however, additional work will be required to establish its sensitivity and specificity for IMNMs relative to other conditions in the histopathologic differential diagnosis (toxic necrotizing myopathies and other IIMs). Advances in neuromuscular disease diagnostics 7. Idiopathic inflammatory myopathies: Myositis-specific antibodies and interferon activation signatures As already mentioned, IIMs are currently divided into four distinct classes - DM, ASM, IMNM, and sIBM - based on the most up-to-date clinicoseropathologic criteria [first comprehensively outlined in a review by Allenbach at al. (2017) and further updated in a recent review by Tanboon and Nishino (2019)]. This classification has been developed based on advances made over the last 5-10 years and is largely (although not entirely) based on the association of individual IIM classes with different myositis-specific antibodies (MSA); while these autoantibodies sometimes directly mediate muscle injury (as already discussed for IMNMs in discovery #6), more often they represent a diagnostically useful epiphenomenon / disease marker. The discovery of MSA has revolutionized IIM diagnostics because of their high specificity, but the sensitivity remains relatively low. For example, two retrospective studies of large European IIM cohorts published in 2019 found that MSA are negative in 40-60% of IIM patients (Betteridge et al., 2019; Montagnese et al., 2019); in these MSA-negative cases (as well as in rare cases with more than one MSA present or in cases for which MSA information is lacking), muscle biopsy remains the cornerstone of IIM diagnosis.
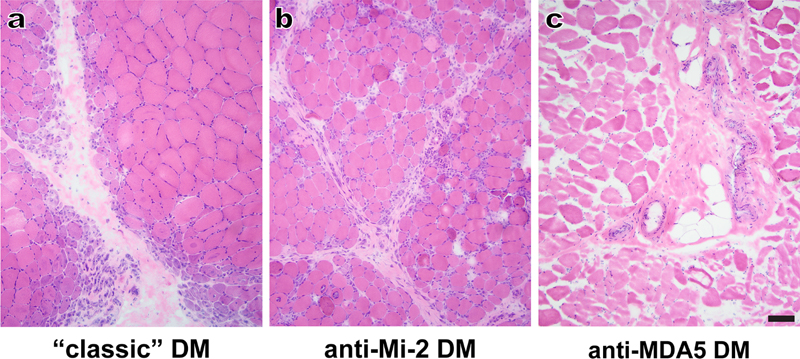
Figure 6. Dermatomyositis (DM) variants: (a) A “classic” DM case shows marked perifascicular fiber atrophy but no significant inflammation in the perimysium or endomysium; the patient’s antibody status is not known, but the overall clinicopathologic findings fit best with the anti-TIF1γ-associated DM. (b) A typical case of anti-Mi-2-associated DM also shows perifascicular atrophy, but could be confused with anti-synthetase syndrome-associated myositis given the presence of perimysial and focally endomysial inflammation, perimysial fragmentation, and patchy necrosis of perifascicular muscle fibers. (c) A case of anti-MDA5-associated DM shows moderate processing artifacts but no significant pathologic changes; the only significant abnormality was mild upregulation of MHC-I complex in all muscle fibers (not shown). For all three cases, images show representative H&E-stained cryosections. Scale bar, 100 µm. Histopathologically, most cases of DM, IMNM, ASM, and sIBM are easily distinguishable [for a review of up-to-date pathologic criteria, see Tanboon and Nishino (2019)]; however, a few entities can create diagnostic difficulties. For example, anti-MDA5-associated subtype of DM generally lacks perifascicular atrophy (a key diagnostic feature of DM; Fig. 6a) and can therefore mimic IMNM or even normal muscle (Fig. 6c), while anti-Mi-2-associated DM shows prominent perimysial inflammation and perifascicular fiber necrosis that can mimic ASM (Fig. 6b). The interferon-induced gene expression signatures associated with different IIM classes, which were defined in two separate studies published in 2019 (Pinal-Fernandez et al., 2019; Rigolet et al., 2019), confirm the validity of the current IIM classification and identify immunohistochemical markers of these interferon responses, which will likely prove diagnostically useful. In particular, both studies have shown that all subtypes of DM (including the histologically atypical cases associated with anti-MDA5 and anti-Mi-2 antibodies) show activation of the type I interferon (INFα/β) pathway and prominent upregulation of IFN1-inducible genes such as MxA (myxovirus resistance protein A), which can be used as an immunohistochemical marker of the INF1 response. In DM, MxA generally shows sarcolemmal staining in perifascicular fibers; the exception is anti-MDA5-associated DM, where MxA staining is scattered or diffuse but not perifascicular (Tanboon and Nishino, 2019). In contrast, both ASM cases (regardless of the nature of the associated anti-synthetase antibody) and sIBM cases show activation of the interferon type II pathway (i.e. upregulation of INFγ-inducible genes); MHC-II, an immunohistochemical marker of INFγ activation, can be used to monitor this pathway in muscle biopsies (Rigolet et al., 2019) and could therefore be useful for distinguishing ASM from anti-Mi-2-DM in challenging cases. [Of note, DM muscle biopsies do show a weak activation of INFγ pathway (Pinal-Fernandez et al., 2019; Rigolet et al., 2019); however, at least for MHC-II proteins that activation is several orders of magnitude lower than in ASM and sIBM biopsies. Thus, it should be possible to optimize MHC-II staining to effectively differentiate between DM and ASM cases.] Finally, both studies found that IMNM biopsies do not exhibit either type I or type II interferon signature, further validating their separation into a separate IIM category (Pinal-Fernandez et al., 2019; Rigolet et al., 2019). Thus, neuromuscular pathology laboratories that see a lot of IIM cases should consider adding MxA and MHC-II immunostains to their stain repertoire, so that these markers can be evaluated in diagnostically challenging IIM cases. What are the sources of INFα/β and INFγ that drive these differential responses? That remains to be definitively established; however, the most likely candidate for the INFα/β source are plasmacytoid dendritic cells, which are increased in DM muscle, while the most likely candidate for the INFγ source are CD8+ T cells, which were observed in a close vicinity of MHC-II-expressing fibers in both ASM and sIBM (Rigolet et al., 2019). 8. Emerging diagnostic techniques The reduced cost and widespread availability of advanced genetic testing techniques [such as disease gene panels and whole exome sequencing (WES)] have led to a dramatic diagnostic improvement for many inherited NMDs. However, even with the aid of these advanced molecular techniques, 50-75% of presumed genetic NMD cases currently remains undiagnosed (Cummings et al., 2017); the underlying reasons include challenges involved in interpretation of the variants of unknown significance and of variants in the genes previously not linked to human disease, the inability of these techniques to detect certain types of genetic alterations (such as structural re-arrangements and copy number variants), and the lack of coverage of intronic and regulatory regions (Gonorazky et al., 2019). In such diagnostically challenging cases, transcriptome analysis (RNA-seq) will likely prove beneficial. Indeed, a recent study has shown that RNA-seq resolved 36% of their panel- and/or WES-negative NMD cases (Gonorazky et al., 2019); an essentially identical diagnostic yield (35%) was reported in a 2017 study that used a different NMD patient cohort (Cummings et al., 2017). Importantly, gene expression is tissue-dependent; thus, the effectiveness of RNA-seq for diagnostic testing hinges on the use of disease-relevant source material. In the context of genetic muscle disease, biopsied muscle represents the ideal specimen, while peripheral blood is inadequate; unmodified skin-derived fibroblasts can be useful for some diseases (such as congenital muscular dystrophies), but the diagnostic yield is substantially enhanced if fibroblasts are transdifferentiated into myotubes in vitro (Gonorazky et al., 2019). Importantly, expression of the muscle-specific transcripts was significantly lower in transdifferentiated myotubes than in the matched muscle biopsy samples for most genes; therefore, muscle biopsies will likely remain the gold standard for diagnostic transcriptomics. However, the transdifferentiation approach may prove useful when muscle biopsy cannot be performed or when accurate diagnosis requires correlation of gene expression patterns between the proband and other family members (Gonorazky et al., 2019). Another approach for improving muscle disease diagnostics is based on the use of automated image-analysis methods and artificial intelligence (AI) tools during muscle biopsy evaluations. To this end, a French group has recently developed a fully automated macro script for Image J that performs muscle fiber measurements (such as total fiber number, type 1 and type 2 fiber proportion, and muscle fiber diameter) that are routinely assessed during muscle biopsy evaluation, but are typically estimated and reported in a semi-quantitative fashion (Reyes-Fernandez et al., 2019). In addition to validating the macro script (which analyzes images from immunofluorescence-stained sections and is available for download as the manuscript supplementary material), this study has used quantitative morphometry to definitively demonstrate that the size of type 2 fibers decreases with age but increases with obesity, an observation that has relevance for routine muscle biopsy interpretation (Reyes-Fernandez et al., 2019). Notably, the authors are in the process of adopting their macro script for use on immunoperoxidase-stained sections, which will make it more routinely applicable to most pathology practices. More provocatively, a study presented at the 2019 World Muscle Society Congress (Kabeya et al., 2019) has shown that a deep neural network model (“AI-based muscle histopathologist”) can use images from H&E-stained slides to differentiate several major muscular dystrophies subtypes better than physicians (although the study abstract does not indicate whether those physicians were expert muscle pathologists). While intriguing, the validity of these results needs to be replicated by future studies. In particular, NMD diagnosis generally requires interpretation of many different stains and integration of clinical, laboratory, and histopathologic findings; thus, it is fairly unlikely that the diagnostic proficiency of an “AI-based muscle histopathologist” will generalize to a broader set of NMDs. At the same time, it is likely that the best diagnostic yield will ultimately be achieved through the combined efforts of AI and human pathologists, as was previously demonstrated for the other fields of medicine that are based on image interpretation (such as radiology). Advances in neuromuscular disease treatment Development of new disease therapies takes decades of work, with clinical trials that span years and report their results thorough multiple papers published over time; as such, it is not entirely appropriate to designate any of the recently approved genetic treatments as a 2019 discovery. At the same time, these new treatments were among of the “hottest” NMD advances in 2019, and therefore had to be included in this review. 9. A success story: Genetic therapies for spinal muscular atrophy Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease that affects the alpha motor neurons in the anterior horn of the spinal cord, resulting in neurogenic muscle atrophy and weakness; the onset of disease varies from the first 6 months of age for the most severe infantile form (SMA1) to the second or third decade for the mildest form (SMA4). SMA is caused by loss-of-function mutations in both copies of SMN1 (survival motor neuron 1) gene, but clinical phenotype is modified by the presence of the paralog gene, SMN2, which yields only a small number of full length transcripts (due to a mutation in the exon 7 that alters gene splicing) and is present in a variable number of copies in different individuals. The first genetic therapy for SMA, an intrathecally delivered antisense oligonucleotide (ASO) called nusinersen, was approved in the US in 2016 and in Europe in 2017 based on the results of clinical trials done with SMA1 and SMA2 infants; additional clinical trials for other SMA subgroups are still ongoing. [For a succinct but comprehensive review of all treatment approaches summarized in this section, see (Vita et al., 2019).] Nusinersen binds to the SMN2 pre-mRNA downstream of exon 7, resulting in an increase in the expression of SMN protein and a significant improvement in motor function and patient survival. Risdiplam, an orally available small molecule therapeutic that also acts as a splicing modifier of SMN2 mRNA, is still in clinical trials; however, based on the promising early results, it has been placed on an accelerated pathway for the approval by EMA (European Medicines Agency). An alternate approach for SMA treatment is to directly replace the defective SMN1 gene; this approach has led to development of AAV9-SMN (also called AVXS-101 or Zolgensma), an AAV9 (adeno-associated virus 9) vector carrying human SMN transgene that was approved for SMA treatment by FDA (Federal Drug Administration) in May 2019 (Al-Zaidy and Mendell, 2019). The availability of multiple treatment options raises many interesting questions that will need to be addressed going forward. Are some of these treatments superior to others? (An indirect comparison between nusinersen and AVXS-101 has been done and suggests that AVXS-101 is more effective (Dabbous et al., 2019); however, that study had significant limitations. Thus, a direct comparison of these two drugs is needed, but will be challenging to perform given the associated cost and ethical considerations.) If two treatments are based on different genetic strategies, would a combination therapy be more effective than either treatment alone? How will treatment efficacy differ depending on the SMA subtype and the age at which the treatment is started? As expected for a neurodegenerative disease, the treatment is more effective if it is initiated before significant neuron loss has occurred (De Vivo et al., 2019); depending on the SMA subtype, this may require perinatal or even prenatal diagnosis. Indeed, a large autopsy study published in 2019 has shown that SMN protein levels in the human spinal cord decline rapidly during development, with a critical window that spans from 3 months before to 3 months after birth (Ramos et al., 2019); this finding highlights the need for development of newborn SMA screening programs, which are already being piloted in several different countries (Dangouloff et al., 2019; Vill et al., 2019). 10. On the horizon: Genetic therapies for inherited myopathies Aside from SMA, the only currently approved genetic therapies for neuromuscular disease are three new drugs for Duchenne muscular dystrophy (DMD), which is a severe progressive X-linked muscle disorder caused by the lack of membrane-associated dystrophin protein; however, based on their rather modest effects to date and on the conditional and inconsistent nature of the approvals that were rendered by FDA and EMA, these treatments have yet to prove their therapeutic utility and are best still considered as being “under development” (Vita et al., 2019). Ataluren is an orally bioavailable drug that binds ribosomal RNA and interferes with recognition of premature stop codons present in 10-15% of DMD patients; it received a conditional approval from EMA in 2014, but the evaluation of long-term outcomes is still ongoing. The other two conditionally approved treatments for DMD are based on the exon-skipping ASO technology; the goal of this therapy is to “trick” the cellular machinery to restore the reading frame by skipping over the mutant exon, resulting in an internally truncated but functional protein. Eteplirsen, which targets the splice-donor region of exon 51 (mutated in 13% of DMD patients), was conditionally approved by FDA in 2016 but failed to receive an approval from EMA (Aartsma-Rus and Goemans, 2019); a phase 3 clinical trial to evaluate its efficacy is still ongoing. Golodirsen (exon 53; 8% of DMD patients) has been approved by FDA in December 2019 (https://www.drugs.com/newdrugs/fda-approves-vyondys-53-golodirsen-duchenne-muscular-dystrophy-dmd-patients-amenable-skipping-exon-5119.html; accessed on 12/26/2019), while clinical trials for casimirsen (exon 45; 8% of DMD patients) and suvodirsen (exon 51) are still ongoing (Vita et al., 2019). Based on the animal model studies, similar exon skipping treatments may also prove valuable for limb-girdle and congenital muscular dystrophies (Hwang and Yokota, 2019). Excitingly, new treatments for other genetic myopathies are also under active development; several different therapeutic strategies are being evaluated through ongoing or planned clinical trials and include AAV vector-based gene replacement therapies (for X-linked myotubular myopathy and Pompe disease), ASO-based therapy aimed at reduction of toxic RNA production (for myotonic dystrophy type 1), chaperone therapy (for Pompe disease), and the second generation enzyme-replacement therapy (also for Pompe disease) (Vita et al., 2019). Initial results that are available to date are promising; thus, with some luck, at least some of these new treatments will prove beneficial enough to be covered in a future update. At the same time, all these new gene therapies are likely to be highly expensive; for example, the list price of nusinersen treatment in the US is $750,000 in the first year and $375,000 / year after that. Thus, an increase in the availability of genetic therapies raises important socio-economic and ethical questions that will have to be addressed before their full benefits can be realized. Disclosure statement The author is a consultant for Audentes Therapeutics; she serves as a member of the muscle biopsy review committee for the ASPIRO clinical trial (NCT03199469), which is evaluating the safety and efficacy of gene transfer in X-linked myotubular myopathy. Acknowledgements I am grateful to Drs. Michael Lawlor, Nigel G. Laing, and Benedikt Schoser for helpful input during the conceptualization stage of this review, to Dr. Emily Sloan for n-SCIPT images, and to Ms. Christine Lin for assistance with figure preparation. This work was supported by the Muscular Dystrophy Association grant MDA514303. References Aartsma-Rus, A., and Goemans, N. (2019). A sequel to the eteplirsen saga: Eteplirsen is approved in the United States but was not approved in Europe. Nucleic Acid Ther 29, 13-15. Allenbach, Y., Benveniste, O., Goebel, H.H., and Stenzel, W. (2017). Integrated classification of inflammatory myopathies. Neuropathol Appl Neurobiol 43, 62-81. Allenbach, Y., Mammen, A.L., Benveniste, O., Stenzel, W., on behalf of the Immune-Mediated Necrotizing Myopathies Working Group (2018). 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord 28, 87-99. Anquetil, C., Salem, J.E., Lebrun-Vignes, B., Johnson, D.B., Mammen, A.L., Stenzel, W., Leonard-Louis, S., Benveniste, O., Moslehi, J.J., and Allenbach, Y. (2018). Immune checkpoint inhibitor-associated myositis. Circulation 138, 743-745. Al-Zaidy, S.A., and Mendell, J.R. (2019). From clinical trials to clinical practice: practical considerations for gene replacement therapy in SMA type 1. Pediatr Neurol 100, 3-11. Arouche-Delaperche, L., Allenbach, Y., Amelin, D., Preusse, C., Mouly, V., Mauhin, W., Tchoupou, G.D., Drouot, L., Boyer, O., Stenzel, W., et al. (2017). Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol 81, 538-548. Bergua, C., Chiavelli, H., Allenbach, Y., Arouche-Delaperche, L., Arnoult, C., Bourdenet, G., Jean, L., Zoubairi, R., Guerout, N., Mahler, M., et al. (2019). In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis 78, 131-139. Betteridge, Z., Tansley, S., Shaddick, G., Chinoy, H., Cooper, R.G., New, R.P., Lilleker, J.B., Vencovsky, J., Chazarain, L., Danko, K., et al. (2019). Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J Autoimmun 101, 48-55. Cummings, B.B., Marshall, J.L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A.R., Bolduc, V., Waddell, L.B., Sandaradura, S.A., O'Grady, G.L., et al. (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 9. Dabbous, O., Maru, B., Jansen, J.P., Lorenzi, M., Cloutier, M., Guerin, A., Pivneva, I., Wu, E.Q., Arjunji, R., Feltner, D., et al. (2019). Survival, motor function, and motor milestones: Comparison of AVXS-101 relative to nusinersen for the treatment of infants with spinal muscular atrophy type 1. Adv Ther 36, 1164-1176. Dangouloff, T., Burghes, A., Tizzano, E.F., Servais, L., and Group, N.S.S. (2019). 244th ENMC international workshop: Newborn screening in spinal muscular atrophy May 10-12, 2019, Hoofdorp, The Netherlands. Neuromuscul Disord, ePub ahead of print. De Ridder, W., Nelson, I., Asselbergh, B., De Paepe, B., Beuvin, M., Ben Yaou, R., Masson, C., Boland, A., Deleuze, J.F., Maisonobe, T., et al. (2019). Muscular dystrophy with arrhythmia caused by loss-of-function mutations in BVES. Neurol Genet 5, e321. De Vivo, D.C., Bertini, E., Swoboda, K.J., Hwu, W.L., Crawford, T.O., Finkel, R.S., Kirschner, J., Kuntz, N.L., Parsons, J.A., Ryan, M.M., et al. (2019). Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord 29, 842-856. de Winter, J.M., Molenaar, J.P., Yuen, M., van der Pijl, R., Shen, S., Conijn, S., van de Locht, M., Willigenburg, M., Bogaards, S.J., van Kleef, E.S., et al. (2020). KBTBD13 is an actin-binding protein that modulates muscle kinetics. J Clin Invest, ePub ahead of print. Dyda, A., Stelzer-Braid, S., Adam, D., Chughtai, A.A., and MacIntyre, C.R. (2018). The association between acute flaccid myelitis (AFM) and Enterovirus D68 (EV-D68) - what is the evidence for causation? Euro Surveill 23. Dzangue-Tchoupou, G., Mariampillai, K., Bolko, L., Amelin, D., Mauhin, W., Corneau, A., Blanc, C., Allenbach, Y., and Benveniste, O. (2019). CD8+T-bet+ cells as a predominant biomarker for inclusion body myositis. Autoimmun Rev 18, 325-333. Fischer, N., Preusse, C., Radke, J., Pehl, D., Allenbach, Y., Schneider, U., Feist, E., von Casteleyn, V., Hahn, K., Ruck, T., et al. (2019). Sequestosome-1 (p62) expression reveals chaperone-assisted selective autophagy in immune-mediated necrotizing myopathies. Brain Pathol, ePub ahead of print. Gonorazky, H.D., Naumenko, S., Ramani, A.K., Nelakuditi, V., Mashouri, P., Wang, P., Kao, D., Ohri, K., Viththiyapaskaran, S., Tarnopolsky, M.A., et al. (2019). Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am J Hum Genet 104, 466-483. Greenberg, S.A., Pinkus, J.L., Kong, S.W., Baecher-Allan, C., Amato, A.A., and Dorfman, D.M. (2019). Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 142, 2590-2604. Hixon, A.M., Yu, G., Leser, J.S., Yagi, S., Clarke, P., Chiu, C.Y., and Tyler, K.L. (2017). A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog 13, e1006199. Hwang, J., and Yokota, T. (2019). Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies. Expert Rev Mol Med 21, e5. Johansen, A., Christensen, S.J., Scheie, D., Hojgaard, J.L.S., and Kondziella, D. (2019). Neuromuscular adverse events associated with anti-PD-1 monoclonal antibodies: Systematic review. Neurology 92, 663-674. Kabeya, Y., Iwamori, T., Yonezawa, S., Takeuchi, Y., Nakano, H., Nagisa, Y., Okubo, M., Inoue, M., Tokumasu, R., Ozawa, I., et al. (2019). AI-based muscle histopathologist can differentiate major muscular dystrophies better than human. Neuromusc Disord 29, S126-127. (Abstract) Knauss, S., Preusse, C., Allenbach, Y., Leonard-Louis, S., Touat, M., Fischer, N., Radbruch, H., Mothes, R., Matyash, V., Bohmerle, W., et al. (2019). PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol Neuroimmunol Neuroinflamm 6, e558. Lornage, X., Schartner, V., Balbueno, I., Biancalana, V., Willis, T., Echaniz-Laguna, A., Scheidecker, S., Quinlivan, R., Fardeau, M., Malfatti, E., et al. (2019). Clinical, histological, and genetic characterization of PYROXD1-related myopathy. Acta Neuropathol Commun 7, 138. Margeta, M. (2019). Autophagy defects in skeletal myopathies. Annu Rev Pathol, ePub ahead of print. Messacar, K., Asturias, E.J., Hixon, A.M., Van Leer-Buter, C., Niesters, H.G.M., Tyler, K.L., Abzug, M.J., and Dominguez, S.R. (2018). Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect Dis 18, e239-e247. Mohn, N., Beutel, G., Gutzmer, R., Ivanyi, P., Satzger, I., and Skripuletz, T. (2019). Neurological immune related adverse events associated with nivolumab, ipilimumab, and pembrolizumab therapy-Review of the literature and future outlook. J Clin Med 8, ePub ahead of print. Montagnese, F., Babacic, H., Eichhorn, P., and Schoser, B. (2019). Evaluating the diagnostic utility of new line immunoassays for myositis antibodies in clinical practice: a retrospective study. J Neurol 266, 1358-1366. Olive, M., Engvall, M., Ravenscroft, G., Cabrera-Serrano, M., Jiao, H., Bortolotti, C.A., Pignataro, M., Lambrughi, M., Jiang, H., Forrest, A.R.R., et al. (2019). Myoglobinopathy is an adult-onset autosomal dominant myopathy with characteristic sarcoplasmic inclusions. Nat Commun 10, 1396. Pinal-Fernandez, I., Casal-Dominguez, M., Derfoul, A., Pak, K., Plotz, P., Miller, F.W., Milisenda, J.C., Grau-Junyent, J.M., Selva-O'Callaghan, A., Paik, J., et al. (2019). Identification of distinctive interferon gene signatures in different types of myositis. Neurology 93, e1193-e1204. Psimaras, D., Velasco, R., Birzu, C., Tamburin, S., Lustberg, M., Bruna, J., and Argyriou, A.A. (2019). Immune checkpoint inhibitors-induced neuromuscular toxicity: from pathogenesis to treatment. J Peripher Nerv Syst 24 Suppl 2, S74-S85. Ramos, D.M., d'Ydewalle, C., Gabbeta, V., Dakka, A., Klein, S.K., Norris, D.A., Matson, J., Taylor, S.J., Zaworski, P.G., Prior, T.W., et al. (2019). Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Invest 129, 4817-4831. Reyes-Fernandez, P.C., Periou, B., Decrouy, X., Relaix, F., and Authier, F.J. (2019). Automated image-analysis method for the quantification of fiber morphometry and fiber type population in human skeletal muscle. Skelet Muscle 9, 15. Rigolet, M., Hou, C., Baba Amer, Y., Aouizerate, J., Periou, B., Gherardi, R.K., Lafuste, P., and Authier, F.J. (2019). Distinct interferon signatures stratify inflammatory and dysimmune myopathies. RMD Open 5, e000811. Ross, J.A., Levy, Y., Ripolone, M., Kolb, J.S., Turmaine, M., Holt, M., Lindqvist, J., Claeys, K.G., Weis, J., Monforte, M., et al. (2019). Impairments in contractility and cytoskeletal organisation cause nuclear defects in nemaline myopathy. Acta Neuropathol 138, 477-495. Schubert, R.D., Hawes, I.A., Ramachandran, P.S., Ramesh, A., Crawford, E.D., Pak, J.E., Wu, W., Cheung, C.K., O'Donovan, B.D., Tato, C.M., et al. (2019). Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat Med 25, 1748-1752. Sloan, E.A., Sampognaro, P.J., Junn, J.C., Chin, C., Jacques, L., Ramachandran, P.S., DeRisi, J.L., Wilson, M.R., Kriegstein, A.R., Bollen, A.W., et al. (2019). Neuroglial stem cell-derived inflammatory pseudotumor (n-SCIPT): clinicopathologic characterization of a novel lesion of the lumbosacral spinal cord and nerve roots following intrathecal allogeneic stem cell intervention. Acta Neuropathol 138, 1103-1106. Tanboon, J., and Nishino, I. (2019). Classification of idiopathic inflammatory myopathies: pathology perspectives. Curr Opin Neurol 32, 704-714. Touat, M., Maisonobe, T., Knauss, S., Ben Hadj Salem, O., Hervier, B., Aure, K., Szwebel, T.A., Kramkimel, N., Lethrosne, C., Bruch, J.F., et al. (2018). Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology 91, e985-e994. Vill, K., Kolbel, H., Schwartz, O., Blaschek, A., Olgemoller, B., Harms, E., Burggraf, S., Roschinger, W., Durner, J., Glaser, D., et al. (2019). One year of newborn screening for SMA - Results of a German pilot project. J Neuromuscul Dis 6, 503-515. Villar-Quiles, R.N., Catervi, F., Cabet, E., Juntas-Morales, R., Genetti, C.A., Gidaro, T., Koparir, A., Yuksel, A., Coppens, S., Deconinck, N., et al. (2019). ASC-1 is a cell cycle regulator associated with severe and mild forms of myopathy. Ann Neurol, ePub ahead of print. Vissing, J., Johnson, K., Topf, A., Nafissi, S., Diaz-Manera, J., French, V.M., Schindler, R.F., Sarathchandra, P., Lokken, N., Rinne, S., et al. (2019). POPDC3 gene variants associate with a new form of limb girdle muscular dystrophy. Ann Neurol 86, 832-843. Vita, G., Vita, G.L., Musumeci, O., Rodolico, C., and Messina, S. (2019). Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurol Sci 40, 671-681.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |