|
|
|
Free Neuropathology 1:5 (2020) |
|
Review |
|
Top ten discoveries of the year: Neurovascular disease |
|
Anna M. Planas |
|
Department of Brain Ischemia and Neurodegeneration, Spanish National Research Council (CSIC), Barcelona, Spain |
|
Corresponding author: |
|
Submitted: 09 January 2020 Accepted: 25 January 2020 Published: 30 January 2020 |
|
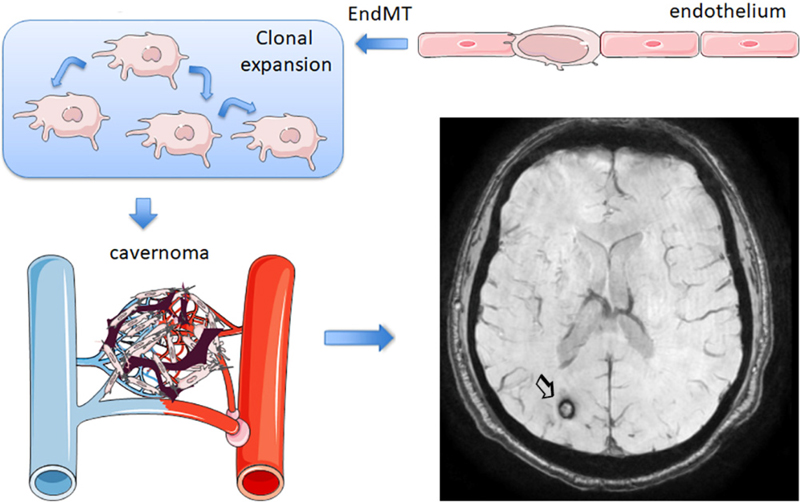
Abstract The aim of this review is to highlight novel findings in 2019 in the area of neurovascular disease. Experimental studies have provided insight into disease development, molecular determinants of pathology, and putative novel therapeutic targets. Studies in genetic experimental models as well as monogenic forms of human cerebrovascular diseases identified pathogenic molecules that may also be relevant to sporadic cases. There have been advances in understanding the development of cerebral cavernous angiomas and arteriovenous malformations, and putative curative treatments have been suggested from experimental models. Key pathogenic pathways involved in vessel calcification and stiffness have also been identified. At the cellular level, studies showed that proper function of endothelial and mural cells, particularly pericytes, is crucial to ensure full endothelial differentiation and blood-brain barrier integrity. Moreover, recent discoveries support the existence of a homeostatic crosstalk between vascular cells and other neural cells, including neurons. Cerebrovascular diseases are strongly associated with inflammation. Beyond pathogenic roles of specific components of the inflammatory response, new discoveries showed interesting interactions between inflammatory molecules and regulators of vascular function. Clinical investigation on cerebrovascular diseases has progressed by combining advanced imaging and genome-wide association studies. Finally, vascular cognitive impairment and dementia are receiving increasing attention. Recent findings suggest that high-salt intake may cause cerebrovascular dysfunction and cognitive impairment independent of hypoperfusion and hypertension. These and other recent reports will surely inspire further research in the field of cerebrovascular disease that will hopefully contribute to improved prevention and treatment. Abbreviations ApoE, Apolipoprotein E; AVM, Arteriovenous malformations; BBB, Blood-brain barrier; CAA, cerebral amyloid angiopathy; CADASIL, Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CBF, Cerebral blood flow; CCM, Cerebral cavernous angiomas; Cdk5, Cyclin-dependent kinase 5; DTI, Diffusion tensor imaging; EndMT, Endothelial-to-mesenchymal transition; GOM, Granular osmiophilic material; iPSCs, induced pluripotent stem cells; Pdgfb, Platelet-derived growth factor subunit β; PFBC, primary familial brain calcification, Fahr’s disease; SAH, Subarachnoid hemorrhage; SMCs, Smooth muscle cells; TIMP3, Tissue inhibitor of metalloproteinase 3; TGFβ, Transforming growth factor-β Introduction In the next pages I will discuss a few topics in the field of cerebrovascular disease that I believe have made advances in 2019 supported by excellent studies covering genetic and sporadic forms of the diseases in humans and experimental animals. 1. Cavernous angiomas originate from clonal expansion of mutated endothelial cells There have been recent advances in understanding the development of cerebral cavernous angiomas (CCM). New results suggest that small angiomas may derive from a clonal expansion of mutant endothelial cells that undergo endothelial-to-mesenchymal transition and express stem-cell like features (Fig. 1). CCM are vascular lesions composed of clusters of capillary-venous blood vessels with thin walls and impaired endothelial tight junctions that usually contain stagnant blood. These abnormal vessels may rupture causing intracranial hemorrhage (Fig. 1) or small subclinical bleeds, which may lead to seizures. Most cases of CCM are spontaneous but 20% of cases may be attributed to autosomal dominant inheritance. The disease is caused by sporadic or inherited mutations in one of the following three genes: CCM1 (KRIT1), CCM2 (MGC4607), or CCM3 (PDCD10). In the affected vessels, endothelial cells suffer endothelial-to-mesenchymal transition (EndMT), express stem cell markers, and undergo loss of typical endothelial cell properties, with increased cell migration, reduced adherence, and increased blood-brain barrier (BBB) permeability. In familial cases, the vascular lesions are discrete although the above genetic mutations affect all cells. Furthermore, heterozygous mice do not develop the disease. The prevalent explanation is the two-hit hypothesis. Endothelial cells carrying a germline mutation in one of the CCM genes need to suffer a second, likely a somatic, mutation that will trigger the pathology. Malinverno et al. (2019) showed that only a few endothelial cells bearing a Ccm3-/- mutation, undergoing EndMT, and expressing progenitor markers, are needed to generate cavernomas because the affected cells undergo clonal expansion. Demonstration of clonal expansion was carried out using an elegant strategy by crossing the ‘R26R-Confetti’ mouse, a multicolor reporter mouse model, first with Cdh5(PAC)-CreERT2 mice for an endothelial specific expression of cre, and then with Ccm3fl/fl mice. Endothelial deletion of Ccm3 was accompanied by color-based clonal lineage tracing. The majority of small vascular lesions were composed of cells of the same color suggesting a clonal origin from proliferation of the affected endothelial cells. This study followed two previous studies (Detter et al., 2018; Manavski et al., 2018) where clonal expansion of mutant endothelial cell in CCM EndMT was also demonstrated using a reporter Confetti strategy. In contrast to the clonal origin of small lesions, large cavernomas are mosaics containing clonally dominant mutant cells and wild type endothelial cells suggesting recruitment of neighboring wild type cells to the cavernous lesion. Those wild type endothelial cells also overexpressed EndMT markers. Transplantation of Ccm3-/- cells into the brain of wild type mice generated abnormal vessels and recruited endothelial cells from the host (Malinverno et al., 2019). By means of several in vitro studies, these authors demonstrated that wild type endothelial cells acquire features of EndMT after contacting Ccm3-/- cells. Furthermore, by using genetic mouse models, Malinverno et al. deleted Ccm3 in endothelial progenitors. They hypothesized that the early steps of cavernoma formation are due to clonal expansion of resident endothelial progenitors. In this way they provided evidence supporting the view that endothelial progenitors suffering Ccm3 mutation trigger cavernoma formation. Hopefully these new discoveries will provide novel avenues to prevent or attenuate CCM.
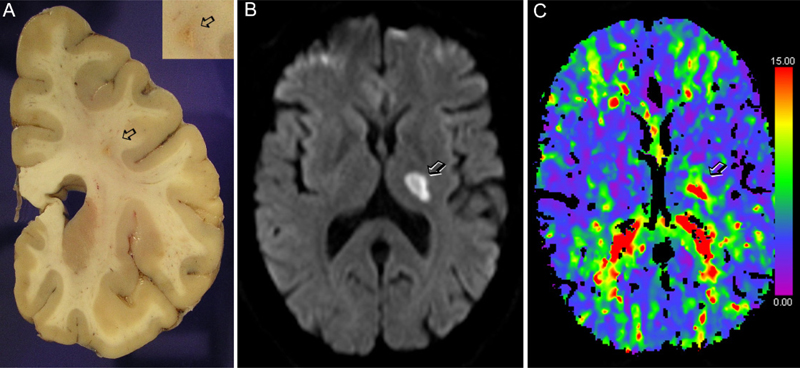
Figure 1. Cavernomas. Schematic representation of cavernoma generation from vessels with mutated CCM genes (see text section 1). According to Malinverno et al., (2019), the process involves endothelial-to-mesenchymal transition (EndMT) and clonal expansion. Susceptibility weighted imaging shows a cavernoma with hemorrhage in the right occipital lobe (arrow). The MRI image was obtained in the Comprehensive Stroke Center of Hospital Clinic of Barcelona. 2. Cerebral arteriovenous malformations: From origin to treatment Failure to achieve full endothelial cell differentiation could underlie the pathology of arteriovenous malformations (AVMs) where recent studies identified β-adrenergic antagonists as putative curative treatments. AVMs involve the formation of arteriovenous shunts amongst other vascular abnormalities including vessel calcifications, and the presence of surrounding macrophages and gliosis. This condition impairs oxygen delivery to the parenchyma and predisposes to vessel rupture and hemorrhage. Alterations in endothelial cell EndMT are associated with this pathology. Yao et al. (2019) recently investigated the presence of EndMT markers in the endothelium of human AVMs. Through complementary strategies they identified the expression of the stem cell marker Sry-box 2 (Sox2) and the mesenchymal marker N-cadherin in lesional endothelial cells, together with a reduction in the expression of typical endothelial markers. The authors also investigated a mouse model of AVMs, the matrix gla protein null (Mgp-/-) mouse, which develops arterial calcifications, enlarged vessels, and arterial-venous shunts. Cerebral endothelial cells of these mice also overexpressed Sox2.They limited Sox2 expression in endothelial cells by generating heterozygous Cdh5-cre/Sox2fl/wt mice with endothelial deletion of Sox2 in heterozygosis. These mice were bred with Mgp-/- mice, which no longer overexpressed Sox2 and, importantly, the pathological vascular features of AVMs were strongly attenuated. Moreover, reduction of Sox2 abolished the expression of EndMT markers. Chip-Seq analysis showed alterations in histone methylation affecting the expression of several genes that participate in stem cell pathways and epithelial-mesenchymal signaling. The analysis identified overexpression of the gene JMJD5 in Mgp-/- cerebral endothelial cells as a downstream target of Sox2. Then, a human brain microvascular endothelial cell line depleted of MGP was generated using CRISP/Cas9 technology. This line showed higher expression of N-cadherin than wild type cells, suggesting EndMT. In addition, these cells overexpressed Sox2 and JMJD5. Sox2 depletion reduced JMJD5 expression. Further experiments suggested that Sox2 also interacts with JMJD5 to regulate EndMT. To identify drugs that could prevent AVMs by inhibiting Sox2, a high-throughput robotic model was generated using a cell line that allowed screening of more than 3,000 products. The system identified a drug called pronethanol, which was shown to effectively reduce Sox2 expression. Furthermore, treatment of Mgp-/- mice with pronethanol for 14 days improved the cerebral vasculature in AVM. Interestingly, pronethanol is a non-selective beta-blocker with the drawback of causing neurological side effects in humans and carcinogenesis in mice. A number of ß-adrenergic drugs were subsequently tested. The results showed that ß-adrenergic antagonists decreased Sox2 and EndMT, and improved lumen formation. This study is a good example of the way mechanistic studies in human tissue, human cells, and animal models lead to the discovery of potential drugs to improve the disease. Previous studies in Mgp-/- mice had shown the contribution of EndMT and the emergence of multipotent cells to the process of ossification and vascular calcification (e.g. Yao et al., 2013). Like in the case of CCMs, failure of brain endothelial cells to acquire or maintain their differentiation state generates aberrant vessels and vascular diseases. 3. Arterial stiffness and calcifications: Molecular insights and consequences Vessel calcification and arterial stiffness are common in the aged population. Arterial stiffness is caused by structural alterations reducing arterial wall distensibility and the arterial capacity to buffer pulsatile cardiac ejection. This condition is epidemiologically associated with hypertension and cognitive decline. Arterial calcification is an important cause of arterial stiffness but the contribution of calcifications to cognitive decline over other associated features is unclear. Muhire et al. (2019) showed in experimental animals that carotid calcification attenuated resting cerebral blood flow (CBF), impaired cerebral autoregulation, and induced cognitive deficits. The study used a mouse model of arterial stiffness induced by direct application of CaCl2 on the carotid artery. The model is characterized by reduced arterial compliance and distensibility, increased thickness of the intima-media, and fragmentation of the internal elastic lamina, without increased systolic blood pressure. Carotid stiffness increased BBB permeability in the hippocampus where the number of collagen IV+ vessels decreased, suggesting a pathogenic role of these calcifications. Regarding the molecular mechanisms underlying vessel calcifications in brain arteries, a recent study pointed to the involvement of osteopontin and TGFβ signaling in the pathogenesis of calcifications (Grand Moursel et al., 2019). Osteopontin is an extracellular glycoprotein with diverse features. It is cleaved by thrombin and cleaved forms can bind to several integrin receptors expressed by immune cells. Given that vascular calcifications are detected in the cerebral cortex in severe forms of cerebral amyloid angiopathy (CAA), the study investigated molecular modulators of arterial calcifications in post-mortem tissue of eight patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D or D-CAA). Calcified vessel walls showed overexpression of medial and abluminal collagen type I. They also overexpressed osteopontin and TGFβ signaling factor phospho-SMAD2/3, and accumulation of these factors preceded overt vessel calcifications. The study found significant positive correlations between the level of osteopontin and phospho-SMAD2/3 and disease severity and suggested the involvement of these molecules in cerebral artery calcifications in CAA. More severe brain vessel calcifications are found in certain rare genetic diseases. Primary familial brain calcification (PFBC) is a heritable disease also termed Fahr’s disease. It is a rare form of brain calcification affecting vessels of the basal ganglia and often manifests with neuropsychiatric symptoms. Zarb et al. (2019) used a mouse model with mutations in Platelet-derived growth factor subunit-β (Pdgfb) gene causing reduced endothelial expression of this factor, resulting in vascular calcifications associated with pericyte loss and BBB alterations. Interestingly, the mouse model recapitulated several behavioral alterations overlapping with PFBC, including sensorimotor deficits, hyperactivity, anxiety and impaired working memory. Cells surrounding brain vessel calcifications expressed osteoblast, osteoclast, and osteocyte markers. The extracellular matrix contained bone matrix proteins. Importantly, blood vessel ossification was also demonstrated in brain tissue from primary familial brain calcification patients. Another consequence of vessel calcifications was activation of microglial and astroglial cells. Given the reported astrogliosis in PFBC patients, astrocytes were investigated in the mouse model. Notably, GFAP+ astrocytes surrounding calcifications expressed oxidative stress and inflammatory neurotoxic markers, such as complement component 3 (C3) and lipocalin 2. 4. Inflammation/innate immunity: Role of complement activation Inflammation is present in most brain diseases affecting the elderly, including cerebrovascular disease. Recently, activation of the complement cascade was shown to play a pathogenic role in subarachnoid hemorrhage. Moreover, interaction between initiator complement component C1q and ApoE was reported. Complement activation has been reported in cerebrovascular diseases and several lines of evidence strongly support the involvement of complement in secondary tissue injury following acute stroke. C3 is amongst key biomarkers associated with increased risk of major disability, mortality and vascular events following ischemic stroke (Zhong et al., 2019). Complement activation is also associated with poor functional outcome following aneurysmal subarachnoid hemorrhage (SAH). A recent study showed that complement components C1q, C3/C3b/iC3B were more abundant in the brain of SAH patients than controls (van Dijk et al., 2019). A single nucleotide polymorphism in C5 correlated with poor outcome in SAH patients, which showed increased levels of C5a in CSF and plasma. In a mouse model of SAH, blocking C5 activation, using mice deficient in C5a or after administration of C5a blocking antibodies, reduced microglial activation and neural cell death. Interestingly, a recent study discovered molecular links between complement and apolipoprotein-E (ApoE). ApoE is a factor associated with Alzheimer’s disease, cerebrovascular diseases, and atherosclerosis (Yin et al., 2019). Human ApoE has four isoforms ApoE, ApoE2, ApoE3, and ApoE4. The ApoE4 allele is an important risk factor for late onset Alzheimer’s disease, whereas several studies indicate that altered ApoE2 could be associated with white matter hyperintensities in small vessel diseases. Yin et al. (2019) showed absence of ApoE-mediated activation of the classical complement cascade in the choroid plexus. Lipid deposits were noticed in choroid plexus of aged ApoE-/- mice regardless of whether they were fed a high fat diet or normal chow. Knockin mice expressing the human form of ApoE4 (ApoE4-KI) also showed lipid deposits in the choroid plexus but only when fed a high fat diet. In contrast, ApoE3-KI mice did not develop those lipid deposits irrespective of diet regimen. Lipid accumulation was paralleled by leukocyte accumulation in the choroid plexus and CSF. Using laser-capture microdissection of choroid plexus followed by differential gene expression profiling, an interferon signature was detected in ApoE4-KI mice. Additionally, upregulation of complement genes was detected in ApoE-/- mice. Accordingly, the choroid plexus of ApoE-/- mice showed accumulation of immunoglobulins, and complement proteins C1q, C3, C4, as well as C5 and anaphylatoxin C3a. Through various in vitro strategies, the study demonstrated that ApoE inhibits the classical pathway of complement activation through Ca2+-dependent high affinity binding to the activated form of C1q, which was elicited by all ApoE isoforms. Notably, the formation of lipid droplets in the choroid plexus was found in human brains with various degrees of Alzheimer’s disease pathology, and higher lipid droplet numbers were found in patients with dementia. Lipid deposits were associated with C1q-ApoE complex. Furthermore, C1q and ApoE colocalized in atherosclerotic plaques. Overall, the study suggests that ApoE may prevent complement activation. Altered inhibitory effects of ApoE on complement due to allelic alterations is proposed as a pathogenic mechanism in Alzheimer’s disease and atherosclerosis. Further studies will hopefully elucidate the role of this new molecular interaction between ApoE and complement in cerebrovascular and neurodegenerative diseases. 5. Cerebral amyloid angiopathy: Microbleeds, microinfarcts and white matter hyperintensities Progress has been made in understanding the neuropathology of CAA as well as the neuropathological correlates of MRI alterations in this condition. CAA can lead to symptomatic lobar intracerebral hemorrhage. MRI of CAA patients may show microbleeds, microinfarcts, and white matter hyperintensities (Reijmer et al., 2015). Van Veluw et al. (2019a) reported that microbleeds and microinfarcts represent different microvascular alterations in CAA. The vessels showing microbleeds had accumulation of amyloid β (Aβ) and fibrin/fibrinogen upstream and downstream of the microbleed, but not in the microbleed region devoid of smooth muscle cells (SMCs). Likewise, SMCs were missing in the regions of microinfarcts, but in this case Aβ was present in the injured core. The study suggested that removal of Aβ in vessels that have lost SMCs might increase the risk of bleeding, which, if confirmed, might potentially have future therapeutic implications. Using high resolution MRI in post-mortem brain tissue with correlative histopathological analyses, Van Veluw et al (2019b) provided relevant information on the neuropathological correlates of neuroimaging alterations. Diffusion tensor imaging (DTI) is the MRI modality best suited to study white matter fiber track orientation and physical features that can assess microstructural integrity of the brain tissue. The post-mortem brain of CAA patients was fixed with 10% formalin for at least 3 weeks. One brain hemisphere was scanned in a 3T MRI scanner for 14 hours. Image analysis included fiber tractography, and 3-mm thick regions of interest were highlighted in parts of the tracts later taken for histology. Formalin fixation alters the MRI diffusion properties, but fractional anisotropy measured in vivo was linearly related with fractional anisotropy acquired ex vivo in the same brains. The tissue was embedded in paraffin and 6-µm thick sections were obtained for histological analyses. The studies found reduced fractional anisotropy in CAA patients compared to controls. In multivariate analysis, fractional anisotropy data was independently associated with tissue rarefaction as assessed with Luxol fast blue & hematoxylin, lower myelin density as assessed with immunohistochemistry against myelin basic protein, and decreased axonal density determined by immunohistochemistry against neurofilament (NF200). Another DTI parameter, mean diffusivity, was increased in CCA patients and was independently associated with myelin density (Van Veluw et al., 2019b). Moreover, increased mean diffusivity in the frontal white matter was associated with CAA severity in the frontal cortex. In contrast, the DTI changes were not related to markers of gliosis or with oligodendrocyte counts. Investigations on the neuropathological correlates of imaging alterations are important because MRI is widely available and can be used to follow the progression of disease. 6. Novel mechanisms underlying CADASIL vasculopathy Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common hereditary monogenic form of cerebral small vessel disease. CADASIL leads to early onset hemorrhagic or ischemic stroke and vascular dementia. Recent findings in CADASIL unraveled novel molecular targets by describing attenuated vascular dysfunction due to inhibitors of endoplasmic reticulum stress and modulators of the cytoskeleton, or due to vascular endothelial growth factor supplementation. CADASIL pathology develops due to mutations in the Notch3 gene which result in dysfunction of vascular SMCs. Notch3 is involved in vasculogenesis and is mainly expressed in SMCs in adults. However, no specific therapies have been developed so far, possibly because there are many aspects of Notch3 function that remain currently unknown. A recent study (Neves et al. 2019) identified molecular targets involved in vascular dysfunction in CADASIL. Notably, the study replicates in a CADASIL mouse model, the TgNotch3R169C mouse, the same findings as in human CADASIL. Samples were obtained from peripheral biopsies of CADASIL patients. Molecular alterations were identified in peripheral arteries despite the fact that CADASIL pathology manifests principally in cerebral vessels. Peripheral vessels of CADASIL patients showed impaired vascular reactivity and altered vascular structure with features suggesting reduced stiffness. Alterations in SMC proliferation, apoptosis, and cytoskeletal disorganization was also noted. Vascular SMCs of CADASIL patients showed increased NOTCH3 signaling, NOTCH3 ectodomain (Notch3ECD) accumulation, and expression of NOTCH3 target genes relative to control patients. The study also showed Nox5 upregulation linked to an exaggerated endoplasmic reticulum stress response and aberrant cytoskeleton-associated protein phosphorylation in CADASIL. Moreover, the study reports that inhibitors of Notch3, Nox5, ER stress, and RhoA/Rho kinase attenuate vascular dysfunction. This finding identifies putative pharmacologic therapeutic targets. Vascular SMCs in CADASIL are surrounded by deposits of granular osmiophilic material (GOM). However, the mechanisms underlying GOM formation and their contribution to disease progression are unknown. A recent study investigated the course of GOM deposit evolution in humanized TgNotch3Arg182Cys mice (Gravesteijn et al., 2019) and in human CADASIL tissue. GOM deposits are electron dense and are composed of Notch3ECD and extracellular matrix proteins, notably tissue inhibitor of metalloproteinase 3 (TIMP3) and clusterin, that accumulate in the basement membrane of vascular SMCs and pericytes. The study proposes a five-stage GOM classification system based on size, morphology, and electron density of the GOM deposits in order to generate a more systematic, uniform, and unbiased way of analyzing GOM accumulations. It appears that new GOM deposits are continuously generated. However, GOM in TgNotch3Arg182Cys mice did not reach the end-stage of GOM accumulation as in human disease. A limitation of the study is that unlike other CADASIL mouse models, the TgNotch3Arg182Cys mouse lacks histologic vascular pathology and functional deficits. The authors suggest that this phenomenon could be attributed to absence of end-stage GOM accumulation in these mice. Beyond mouse models, the cellular CADASIL pathology has been investigated in patient-specific vascular mural cells generated from induced pluripotent stem cells (iPSCs) obtained from skin biopsies of CADASIL patients (Kelleher et al., 2019). This study showed alterations in endothelial capillary structures caused by the aberrant NOTCH3 expression in mural cells, and corrective effects of vascular endothelial growth factor supplementation. These novel advances are steps toward the identification of novel therapeutic targets in this monogenic form of small vessel disease. 7. Genetic variants in small vessel disease and molecular target discovery In addition to studies examining monogenic forms of small vessel disease, there are efforts to study genetic variation in large cohorts of patients to identify genes involved in sporadic small vessel disease. Genome-wide association studies correlating variants with MRI features of sporadic small vessel disease may lead to the identification of new putative pharmacologic targets. Small vessel disease is often diagnosed on MRI, with characteristic findings including white matter hyperintensities and signs of lacunar infarcts (Fig. 2). Some recent studies investigated the relationship between genetics and white matter hyperintensities in small vessel disease. Traylor et al (2019) carried out a genome-wide association meta-analysis of white matter hyperintensity volumes in 11,226 subjects, including 2,797 stroke patients. The study identified a locus at genome-wide significance in an intron of Pleckstrin Homology and RhoGEF Domain-Containing Family G Member 1 gene (PLEKHG1) associated with white matter hyperintensities. The association was validated in an independent cohort where this polymorphism was related with ischemic strokes. The strongest association was found with small vessel stroke. PLEKHG1 plays a role in vascular endothelial cell reorientation in response to mechanical stress. Therefore, the polymorphism in this gene could generate some form of vascular alteration leading to the development of white matter hyperintensities. This study also validated 2 previously identified genes EFEMP1 and TRIM47/TRIM65. According to current knowledge, EFEMP1 encodes the Fibulin3 protein, which is an extracellular matrix glycoprotein that inhibits TIMP3. TIMP3 contributes to cerebrovascular dysfunction in CADASIL through accumulation in the vascular extracellular matrix. Therefore, there seems to be some concordance between genetic alterations in sporadic small vessel disease and the monogenic form of CADASIL. Based on the concept that polymorphisms in genes involved in monogenic forms of small vessel disease may have some contribution in the sporadic form of the disease, Mishra et al. (2019) carried out the first whole exome sequencing study on MRI white matter hyperintensities in small vessel disease. This work used a composite extreme phenotype study focused on candidate genes with mutations causing Mendelian heritable forms of small vessel disease, i.e. NOTCH3, HTRA1, COL4A1, COL4A2 and TREX1. The study identified significant associations of gene variants in HTRA1 and NOTCH3 with the development of white matter hyperintensities. Overall the study demonstrates shared mechanisms between monogenic forms and multifactorial forms of small vessel disease. The study suggested that the risk HTRA1 allele reduced HTRA1 expression. Mutations in HTRA1 are associated with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, CARASIL, a condition inherited in an autosomal recessive pattern. HTRA1 is a serine-protease involved in cleaving extracellular matrix proteins, including Fibulin3, and regulating important signaling pathways such as the insulin growth factor and TGFß pathway, which suffers an inhibitory effect. Notably, HTRA1 is differentially expressed in astrocytes (Chen et al., 2018) suggesting that dysfunction of this pathway may alter astrocyte function leading to compromised integrity of the neurovascular unit and the BBB. Hopefully additional studies using iPSC from patients and genetically modified animal models further investigating molecular targets identified in prior genetic studies will lead to the discovery of treatments to prevent and attenuate the progression of small vessel disease.
Figure 2. Lacunar infarcts typical of small vessel disease. A) Human brain tissue showing a lacunar infarct (arrow) in the centrum semiovale. Image obtained by Dr. Iban Aldecoa, Neurological Tissue Bank of the Biobank of Hospital Clínic - IDIBAPS - University of Barcelona. B) MRI showing a thalamic lacunar infarct (arrow) and C) corresponding CT-perfusion time-to-drain (TTD) image obtained in the acute phase showing a delay in blood flow in the lacunar infarction (arrow). Images in B and C were obtained in the Comprehensive Stroke Center of Hospital Clinic of Barcelona. 8. Cerebral hypoperfusion and vascular cognitive impairment: Risks of high-salt diet Chronic cerebral hypoperfusion has been associated with diffuse white matter damage and cognitive impairment of vascular origin. Vascular pathology is also frequently observed in neurodegenerative dementias. Hase et al. (2019) investigated the histopathologic features of white matter capillaries by studying approximately 0.7 million capillary profiles from the frontal lobe white matter of 153 subjects with different dementia types including Alzheimer’s disease, dementia with Lewy bodies, dementia in Parkinson’s disease, vascular dementia, mixed dementia, post-stroke dementia and post-stroke no dementia. Control samples were examined from old and young individuals devoid of brain pathology. All dementias showed higher vascular pathology scores than corresponding age-matched controls. In relation to these controls, white matter lesion scores were also higher in all dementias, with the highest values corresponding to vascular dementia and post-stroke dementia. The two latter conditions showed the highest abundance of string vessels, which were observed to a lesser extent in the other dementia types. The density of COL4+ and GLUT1+ capillaries was lower in the white matter than in the cortex and was also lower in dementia patients versus aged controls, with the exception of individuals that had suffered a stroke. The capillary width in the white matter was 31% greater than in the cortex. Notably, white matter capillaries were wider in all dementias compared to all controls irrespective of age. Moreover, there was a moderate positive correlation between capillary width and white matter lesion scores. These results indicate adaptive compensatory mechanisms for chronic hypoperfusion in aging-related dementias. An emerging risk factor recently recognized as a driver of cerebral vascular dysfunction and hypoperfusion is high-salt intake (Faraco et al., 2018). Interestingly, the underlying mechanism is not dependent on salt-induced hypertension but on a complex innate immune response mediated by alterations in the gut immune system (Faraco et al., 2018). Last year, a study by the same team reported that chronic dietary high-salt intake in mice promotes cognitive impairment by inducing phosphorylation of the microtubule-associated protein tau rather than by insufficient cerebral perfusion (Faraco et al., 2019). The presence of hyperphosphorylated tau is one of the hallmarks of Alzheimer’s disease that has also been reported in patients with vascular cognitive impairment. Mice exposed to high-salt diet for several months developed signs of cognitive impairment that correlated with the phosphorylation and aggregation of tau in the brain tissue. Through different experimental strategies, the study demonstrated that high-salt diet impaired the production of endothelial nitric oxide following denitrosylation of calpain. Deficient nitric oxide generation activated cyclin-dependent kinase 5 (Cdk5) which caused tau phosphorylation. Although these important findings were obtained in mice with unrealistically high levels of salt intake, the study identifies a causal relation between high-salt intake, vascular dysfunction, tau pathology, and cognitive impairment independent of cerebral hypoperfusion. 9. Critical role of pericytes in BBB function and more Pericytes are mural cells surrounding capillaries that play essential functions in development and maintenance of BBB integrity. Pericytes facilitate molecular pathways critical in preventing vascular malformations, and it appears that these cells can generate factors that directly support neuronal viability. Recent studies have provided valuable information regarding the biology of pericytes as well as their role in vascular function. Using Pdgfrb-CreERT2 mice with Cre expression induced in mural cells by tamoxifen administration, Diéguez-Hurtado et al. (2019) deleted the expression of the Rbpj gene encoding a transcriptional regulator of the Notch signaling pathway. Ablation of Rbpj at early postnatal stages caused brain hemorrhages, AVMs at the pial surface, endothelial cell hyperplasia and apoptosis, enlargement of the subendothelial basement membrane, and reduced blood flow. These changes were accompanied by astroglial and microglia reactions, inflammation, edema, and some neuronal loss around severely affected blood vessels. Rbpj deletion upregulated genes of the TGFß pathway. The study provides evidence showing that mice with Rbpj gene deficiency in mural cells developed signs of CCMs. Interestingly, when Rbpj was deleted in mural cells of adult mice, the above phenotype was no longer observed. However, induction of permanent brain ischemia in these mice caused increased edema, larger brain lesion volumes, and exacerbated inflammation and vascular abnormalities. Beyond the critical role of pericytes in brain vascular function, Nikolakopoulou et al. (2019) elucidated the role of pericytes in neuronal survival under conditions of vascular dysfunction. A mouse pericyte inducible cre-line was generated using a double gene promoter strategy including both Pdgfrb and Cspg4 promoters to increase the specificity of Cre expression in pericytes over other cells. This line was crossed with mice expressing inducible human diphtheria toxin receptor under control of an upstream loxP-flanked STOP sequence. Injection of tamoxifen induced DTR expression in pericytes which then became susceptible to diphtheria toxin. Ablation of pericytes in adult mice reduced tight junction proteins zonula occludens and occludin, reduced adherens junction VE-cadherin on cortical and hippocampal capillaries, decreased CBF, and increased BBB breakdown. Pericyte loss was followed by vasogenic edema, confirming the involvement of pericytes in maintaining adult BBB function. Furthermore, neuron loss in the cortex and hippocampus as well as behavioral deficits were detected several days after pericyte ablation. A brain-specific pericyte-secreted growth factor termed pleiotropin was hypothesized to prevent neuronal loss. In vitro, pleiotropin was able to protect neurons under hypoxic conditions. In vivo, silencing pleiotropin expression was not sufficient to cause neuronal loss. However, ischemia or excitotoxic lesions in mice with silenced pleiotropin expression caused larger infarctions and more neuronal degeneration. The study concluded that pericyte-dependent pleiotropin-mediated neurotrophic support promotes neuronal survival under circulatory stress. The expedient neuronal loss found in this model of pericyte ablation is intriguing because it was not found in previous pericyte-deficient models. The findings could be relevant to other neurodegenerative conditions if the dependence of neuronal survival on neurotrophic factors provided by pericytes under vascular stress is confirmed. 10. Molecules derived from the vascular endothelium impact neuronal function In addition to pericytes, endothelial cells seem to signal to neurons. Tan et al. (2019) showed that the endothelium secretes semaphorin 3G (Sema3G) which regulates synaptic structure and plasticity in hippocampal neurons through actions on the neuronal neuropilin-2/PlexinA4 holoreceptor. This study suggests a direct link between endothelial-derived factors and neuronal function beyond the supply of oxygen and nutrients through blood flow and proposes a molecular mechanism by which endothelial dysfunction can alter neuronal circuits and cognition. The study shows that Sema3G increases excitatory synapse density via neuropilin-2/PlexinA4 signaling and activation of Rac1. To determine whether Sema3G was derived from endothelial cells, mice bearing a Sema3G deletion selectively in endothelial cells were obtained by crossing Cdh5-Cre mice with floxed Sema3G mice. These mice showed less dendritic spine density in the CA1 and impaired memory. Nonetheless, Sema3G deletion with this strategy would cause Sema3G deficiency during developmental stages which may cause indirect alterations and compensatory changes. Generation of tamoxifen inducible Cdh5-creERT2/Sema3Gfl/fl mice allowed deleting Sema3G in the endothelium of adult mice. This inducible deletion significantly impaired hippocampal long-term potentiation frequency and altered miniature excitatory post-synaptic currents in CA1 pyramidal neurons. Furthermore, Sema3G overexpression improved spine density loss and cognitive defects in Cdh5-creERT2/Sema3Gfl/fl mice. The results suggest that endothelium-derived Sema3G is required for normal hippocampal synaptic plasticity in adulthood. This study raises many questions regarding the mode of transit of Sema3G from the vascular endothelium to the neurons through the vascular and astrocytic basal laminae and astrocytic end-feet, and whether the source of Sema3G is the capillary endothelium. The discovery of molecular interactions between endothelial cells and neurons suggest that endothelial cells influence neuronal function. Therefore, alterations in the vascular endothelium might exert a direct effect on dendritic spine function and contribute to cognitive decline and memory loss. Other recent studies further support the view that molecules generated by the cerebrovascular endothelium have a direct impact on neuronal function. Liu et al. (2020) showed that mice with a selective deletion of Cdk5 gene show a higher endothelial chemokine Cxcl1 expression. Therefore, dysregulation of Cdk5 expression and/or function seems to be critical in different forms of cerebrovascular alterations. Endothelial Cdk5 knockout mice showed astrogliosis in the hippocampus and weakened astrocytic glutamate current mediated by the astroglial glutamate transporter GLT1. The effects on astroglia were mediated by endothelial-derived Cxcl1 acting on the astrocytic receptor Cxcr2. These alterations had consequences for neurons since they reduced glutamate uptake and increased the excitability of hippocampal pyramidal neurons causing seizures in mice with endothelial-deficient Cdk5. Although current evidence is still scarce and based on experimental animal studies, the possibility that vascular endothelial cells generate molecules that signal to neurons and astrocytes and govern neuronal function warrants future studies. Conclusion The above studies discovered contributing factors to neurovascular pathology. Adequate CBF supply and BBB integrity require homeostasis in the cellular and extracellular environment around blood vessels. Moreo-ver, the concept is emerging that pericytes and endothe-lial cells can signal to neurons by releasing certain specif-ic molecular cues. Ideally, the novel knowledge of genet-ic risk factors, molecular mechanisms, cell and system interactions, as well as pathologic and imaging hallmarks in cerebrovascular diseases should translate into curative treatments that are currently unavailable. Acknowledgements Supported by the Spanish Ministry of Science, Innovation and Universities (SAF2017-87459-R). Note I apologize to the authors that have provided highly valuable contributions in the field in 2019 but could not be cited here due to limitations in the number of selected references. References Chen J, Van Gulden S, McGuire TL, Fleming AC, Oka C, Kessler JA, Peng CY. BMP-Responsive Protease HtrA1 Is Differentially Expressed in Astrocytes and Regulates Astrocytic Development and Injury Response. J Neurosci. 2018; 38:3840-3857. doi: 10.1523/JNEUROSCI.2031-17.2018. Detter MR, Snellings DA, Marchuk DA. Cerebral Cavernous Malformations Develop Through Clonal Expansion of Mutant Endothelial Cells. Circ Res. 2018; 123(10):1143-1151. doi: 10.1161/CIRCRESAHA.118.313970. Diéguez-Hurtado R, Kato K, Giaimo BD, Nieminen-Kelhä M, Arf H, Ferrante F, Bartkuhn M, Zimmermann T, Bixel MG, Eilken HM, Adams S, Borggrefe T, Vajkoczy P, Adams RH. Loss of the transcription factor RBPJ induces disease-promoting properties in brain pericytes. Nat Commun. 2019; 10(1):2817. doi: 10.1038/s41467-019-10643-w. Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, Buendia I, Santisteban MM, Segarra SG, Koizumi K, Sugiyama Y, Murphy M, Voss H, Anrather J, Iadecola C. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018; 21(2):240-249. doi: 10.1038/s41593-017-0059-z. Faraco G, Hochrainer K, Segarra SG, Schaeffer S, Santisteban MM, Menon A, Jiang H, Holtzman DM, Anrather J, Iadecola C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature. 2019 Oct;574(7780):686-690. doi: 10.1038/s41586-019-1688-z. Grand Moursel L, van der Graaf LM, Bulk M, van Roon-Mom WMC, van der Weerd L. Osteopontin and phospho-SMAD2/3 are associated with calcification of vessels in D-CAA, an hereditary cerebral amyloid angiopathy. Brain Pathol. 2019; 29(6):793-802. doi: 10.1111/bpa.12721. Gravesteijn G, Munting LP, Overzier M, Mulder AA, Hegeman I, Derieppe M, Koster AJ, van Duinen SG, Meijer OC, Aartsma-Rus A, van der Weerd L, Jost CR, van den Maagdenberg AMJM, Rutten JW, Lesnik Oberstein SAJ. Progression and Classification of Granular Osmiophilic Material (GOM) Deposits in Functionally Characterized Human NOTCH3 Transgenic Mice. Transl Stroke Res. 2019 Oct 30. doi: 10.1007/s12975-019-00742-7. Hase Y, Ding R, Harrison G, Hawthorne E, King A, Gettings S, Platten C, Stevenson W, Craggs LJL, Kalaria RN. White matter capillaries in vascular and neurodegenerative dementias. Acta Neuropathol Commun. 2019; 7(1):16. doi: 10.1186/s40478-019-0666-x. Kelleher J, Dickinson A, Cain S, Hu Y, Bates N, Harvey A, Ren J, Zhang W, Moreton FC, Muir KW, Ward C, Touyz RM, Sharma P, Xu Q, Kimber SJ, Wang T. Patient-Specific iPSC Model of a Genetic Vascular Dementia Syndrome Reveals Failure of Mural Cells to Stabilize Capillary Structures. Stem Cell Reports. 2019; 13(5):817-831. doi: 10.1016/j.stemcr.2019.10.004. Liu XX, Yang L, Shao LX, He Y, Wu G, Bao YH, Lu NN, Gong DM, Lu YP, Cui TT, Sun NH, Chen DY, Shi WX, Fukunaga K, Chen HS, Chen Z, Han F, Lu YM. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J Exp Med. 2020; 217(1). pii: e20180992. doi: 10.1084/jem.20180992. Malinverno M, Maderna C, Abu Taha A, Corada M, Orsenigo F, Valentino M, Pisati F, Fusco C, Graziano P, Giannotta M, Yu QC, Zeng YA, Lampugnani MG, Magnusson PU, Dejana E. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun. 2019; 10(1):2761. doi: 10.1038/s41467-019-10707-x. Manavski Y, Lucas T, Glaser SF, Dorsheimer L, Günther S, Braun T, Rieger MA, Zeiher AM, Boon RA, Dimmeler S. Clonal Expansion of Endothelial Cells Contributes to Ischemia-Induced Neovascularization. Circ Res. 2018; 122(5):670-677. doi: 10.1161/CIRCRESAHA.117.312310. Mishra A, Chauhan G, Violleau MH, Vojinovic D, Jian X, Bis JC, Li S, Saba Y, Grenier-Boley B, Yang Q, Bartz TM, Hofer E, Soumaré A, Peng F, Duperron MG, Foglio M, Mosley TH, Schmidt R, Psaty BM, Launer LJ, Boerwinkle E, Zhu Y, Mazoyer B, Lathrop M, Bellenguez C, Van Duijn CM, Ikram MA, Schmidt H, Longstreth WT, Fornage M, Seshadri S, Joutel A, Tzourio C, Debette S. Association of variants in HTRA1 and NOTCH3 with MRI-defined extremes of cerebral small vessel disease in older subjects. Brain. 2019; 142(4):1009-1023. doi: 10.1093/brain/awz024. Muhire G, Iulita MF, Vallerand D, Youwakim J, Gratuze M, Petry FR, Planel E, Ferland G, Girouard H. Arterial Stiffness Due to Carotid Calcification Disrupts Cerebral Blood Flow Regulation and Leads to Cognitive Deficits. J Am Heart Assoc. 2019; 8(9):e011630. doi: 10.1161/JAHA.118.011630. Neves KB, Harvey AP, Moreton F, Montezano AC, Rios FJ, Alves-Lopes R, Nguyen Dinh Cat A, Rocchicciolli P, Delles C, Joutel A, Muir K, Touyz RM. ER stress and Rho kinase activation underlie the vasculopathy of CADASIL. JCI Insight. 2019; 4(23). pii: 131344. doi: 10.1172/jci.insight.131344. Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, Sagare AP, Lazic D, Sweeney MD, Kong P, Wang M, Owens NC, Lawson EJ, Xie X, Zhao Z, Zlokovic BV. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019; 22(7):1089-1098. doi: 10.1038/s41593-019-0434-z. Reijmer YD, Fotiadis P, Martinez-Ramirez S, Salat DH, Schultz A, Shoamanesh A, Ayres AM, Vashkevich A, Rosas D, Schwab K, Leemans A, Biessels GJ, Rosand J, Johnson KA, Viswanathan A, Gurol ME, Greenberg SM. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain. 2015;138(Pt 1):179-88. doi: 10.1093/brain/awu316. Tan C, Lu NN, Wang CK, Chen DY, Sun NH, Lyu H, Körbelin J, Shi WX, Fukunaga K, Lu YM, Han F. Endothelium-Derived Semaphorin 3G Regulates Hippocampal Synaptic Structure and Plasticity via Neuropilin-2/PlexinA4. Neuron. 2019; 101(5):920-937.e13. doi: 10.1016/j.neuron.2018.12.036. Traylor M, Tozer DJ, Croall ID, Lisiecka-Ford DM, Olorunda AO, Boncoraglio G, Dichgans M, Lemmens R, Rosand J, Rost NS, Rothwell PM, Sudlow CLM, Thijs V, Rutten-Jacobs L, Markus HS; International Stroke Genetics Consortium. Genetic variation in PLEKHG1 is associated with white matter hyperintensities (n = 11,226). Neurology. 2019; 92(8):e749-e757. doi: 10.1212/WNL.0000000000006952. van Dijk BJ, Meijers JCM, Kloek AT, Knaup VL, Rinkel GJE, Morgan BP, van der Kamp MJ, Osuka K, Aronica E, Ruigrok YM, van de Beek D, Brouwer M, Pekna M, Hol EM, Vergouwen MDI. Complement C5 Contributes to Brain Injury After Subarachnoid Hemorrhage. Transl Stroke Res. 2019 Dec 6. doi: 10.1007/s12975-019-00757-0. van Veluw SJ, Reijmer YD, van der Kouwe AJ, Charidimou A, Riley GA, Leemans A, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Histopathology of diffusion imaging abnormalities in cerebral amyloid angiopathy. Neurology. 2019b; 92(9):e933-e943. doi: 10.1212/WNL.0000000000007005. van Veluw SJ, Scherlek AA, Freeze WM, Ter Telgte A, van der Kouwe AJ, Bacskai BJ, Frosch MP, Greenberg SM. Different microvascular alterations underlie microbleeds and microinfarcts. Ann Neurol. 2019a; 86(2):279-292. doi: 10.1002/ana.25512. Yao J, Wu X, Zhang D, Wang L, Zhang L, Reynolds EX, Hernandez C, Boström KI, Yao Y. Elevated endothelial Sox2 causes lumen disruption and cerebral arteriovenous malformations. J Clin Invest. 2019; 129(8):3121-3133. doi: 10.1172/JCI125965. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Boström KI. A role for the endothelium in vascular calcification. Circ Res. 2013; 113(5):495-504. doi: 10.1161/CIRCRESAHA.113.301792. Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, Nietzsche S, Westermann M, Peng L, Hu D, Bontha SV, Srikakulapu P, Beer M, Megens RTA, Steffens S, Hildner M, Halder LD, Eckstein HH, Pelisek J, Herms J, Roeber S, Arzberger T, Borodovsky A, Habenicht L, Binder CJ, Weber C, Zipfel PF, Skerka C, Habenicht AJR. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):496-506. doi: 10.1038/s41591-018-0336-8. Zarb Y, Weber-Stadlbauer U, Kirschenbaum D, Kindler DR, Richetto J, Keller D, Rademakers R, Dickson DW, Pasch A, Byzova T, Nahar K, Voigt FF, Helmchen F, Boss A, Aguzzi A, Klohs J, Keller A. Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain. 2019; 142(4):885-902. doi: 10.1093/brain/awz032. Zhong C, Zhu Z, Wang A, Xu T, Bu X, Peng H, Yang J, Han L, Chen J, Xu T, Peng Y, Wang J, Li Q, Ju Z, Geng D, He J, Zhang Y. Multiple biomarkers covering distinct pathways for predicting outcomes after ischemic stroke. Neurology. 2019; 92(4):e295-e304. doi: 10.1212/WNL.0000000000006717.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |